Jan 30, 2023 4:45:36 PM
By Elizabeth Denton

Peptides, by nature, are composed of amino acids with potentially ionizable chemical moieties. The ionization state of any of these moieties can significantly impact the peptide’s chromatographic behavior, both in terms of peak shape and retention by the solid support. Peptide purification by reversed-phase chromatography, however, almost exclusively includes an acidic additive to the mobile phase solvents, maintaining the solution at a pH of 2-3 throughout the purification cycle. But have you ever considered trying an alternative additive in the mobile phase to improve your purification results?
In the following post I discuss the impact of mobile phase pH in the purification of oxytocin (CYIQNCPLG-NH2), a 9-amino acid peptide that requires disulfide bond-mediated cyclization for its biological activity.
In the case of oxytocin, it is critical to separate the linear from cyclized peptide if any biological assays are to be completed in the future. As with all other peptides I’ve synthesized, I analyzed the crude peptide mixtures by analytical HPLC-MS containing formic acid in the mobile phase solvents. The addition of 0.1% formic acid attains a pH of 2.5-2.7 under these conditions. I was pleased to see during this analysis that the linear and cyclized peptides are well resolved, Figure 1.
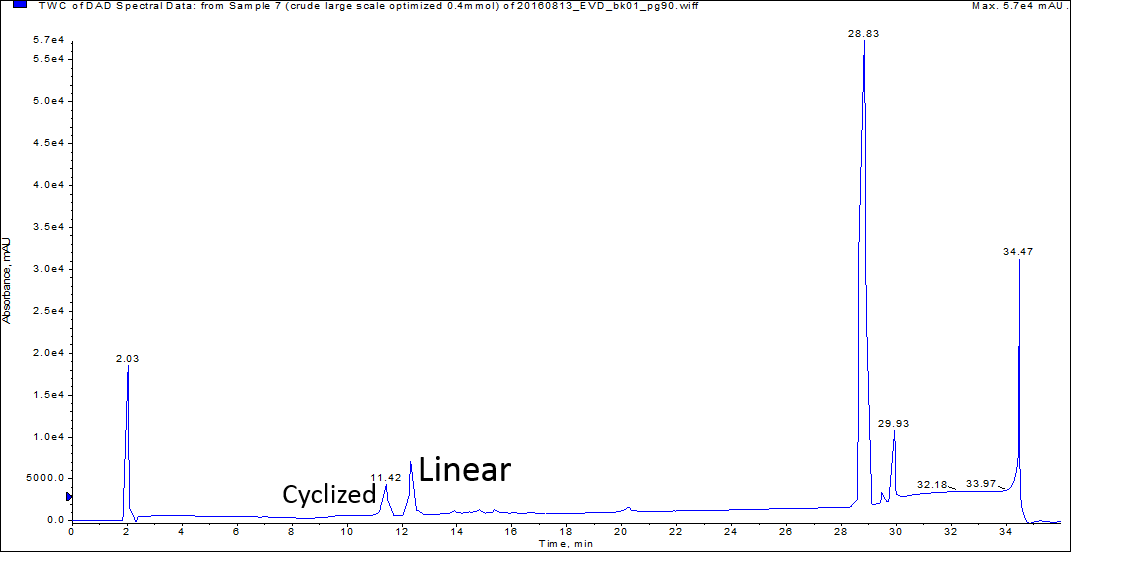 Figure 1: Crude analytical HPLC of a peptide sample containing both linear and disulfide-mediated cyclized oxytocin.
Figure 1: Crude analytical HPLC of a peptide sample containing both linear and disulfide-mediated cyclized oxytocin.
Unfortunately though, after many attempts to separate these two peptides by varying the gradient with a Biotage® SNAP Ultra C18 cartridge, I resigned myself to the fact that flash column chromatography is not selective enough to separate the linear from cyclized oxytocin peptides, Figure 2.
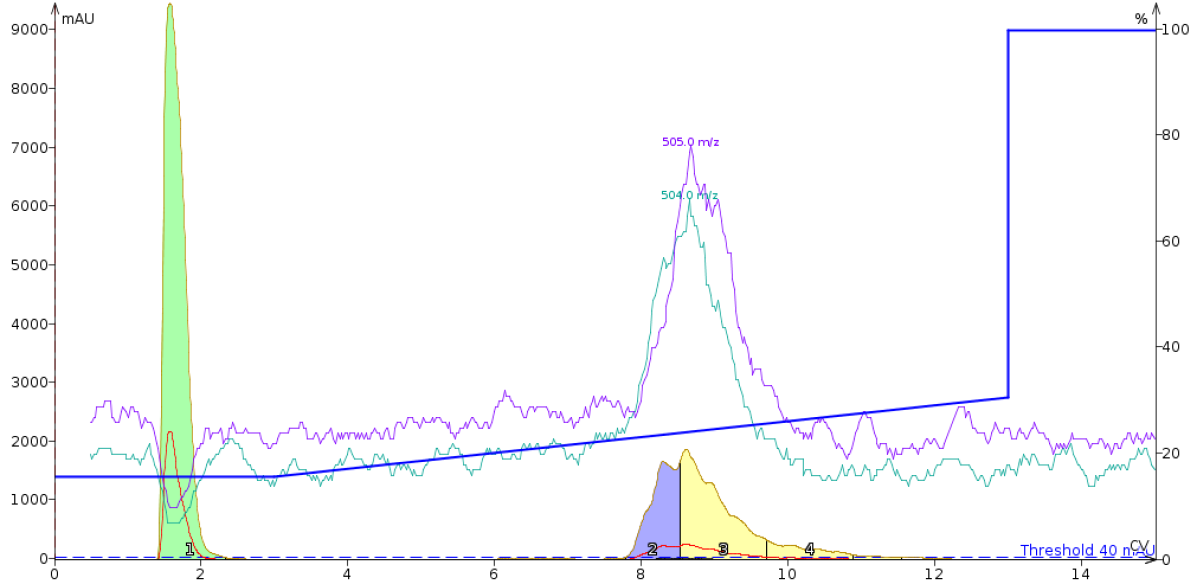
Figure 2: Optimized flash column chromatography gradient to separate linear from cyclized oxytocin dissolved in DMSO. The gradient runs from 15% to 30% acetonitrile with mobile phase solvents containing 0.1% TFA.
Rather than give up on flash column chromatography and return to HPLC (I only have an analytical system with no fraction collector), I thought about the sequence of oxytocin and any features that may chromatographically differentiate the linear from cyclized peptide. Glaring back at me were two reduced Cys sidechains (pKa about 8.37) that could be ionized under basic conditions. After some searching for mobile phase additives that would achieve pH>9 but still maintain compatiblity with LCMS, I settled on 0.1% ammonium hydroxide (pH ~ 10.7). Ideally, the pH should be 1-2 units above the pKa to ensure complete deprotonation of the Cys sulfur atoms and quality chromatography.
I was shocked by the dramatic difference in chromatography I observed after injecting oxytocin, dissolved in DMSO, with the basic mobile phase, Figure 3. Please note that this sample was monitored with negative ionization mode on the mass spectrometer.
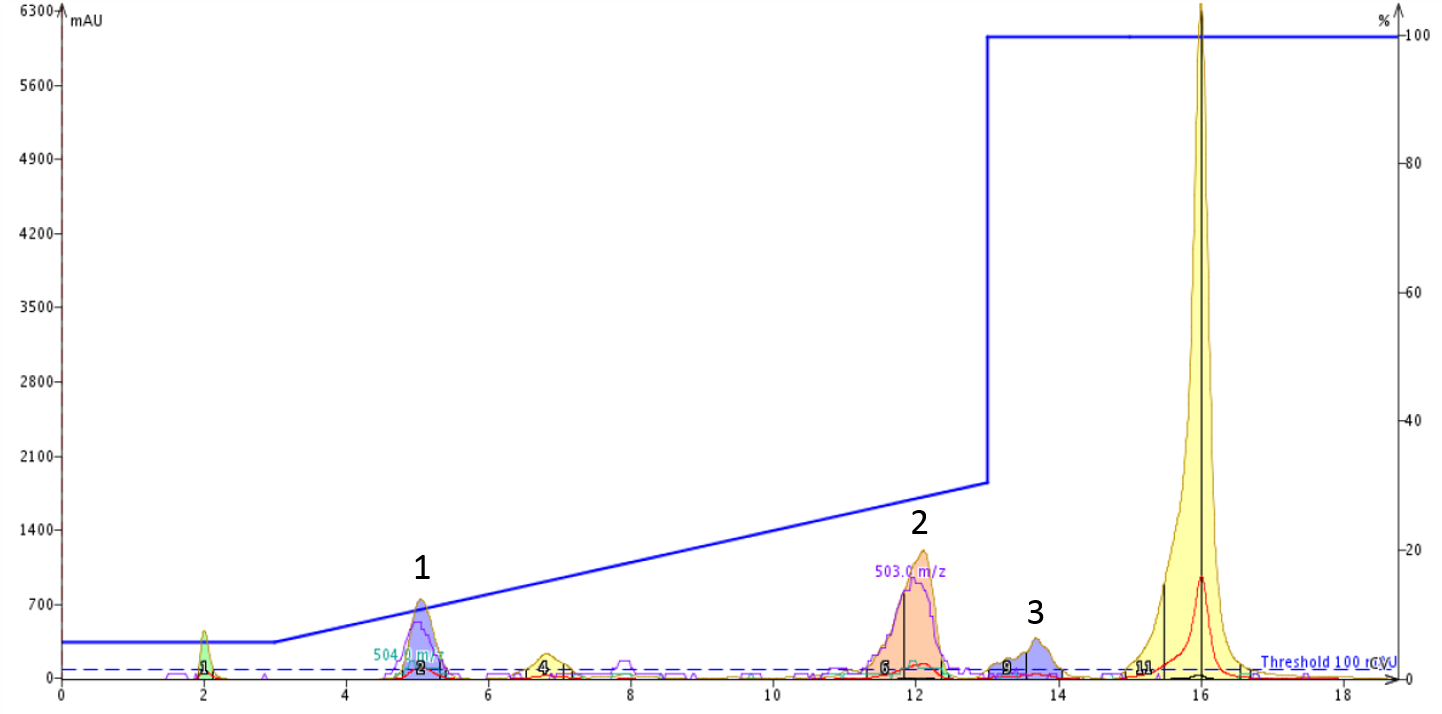
Figure 3: Optimized flash column chromatography gradient to separate linear from cyclized oxytocin dissolved in DMSO. The gradient runs from 5% to 30% acetonitrile with mobile phase solvents containing 0.1% ammonium hydroxide.
I analyzed fractions within each of the three major peaks by analytical HPLC-MS and found that the cyclized peptide was exclusively contained within the peak 1 and that the linear peptide was contained in both peaks 2 and 3 (I checked, nothing with peptidic character is present in the large peak at the end of the run). I would normally expect the linear, deprotonated, peptide to elute from the column early, but that is not what was observed here. Given the extreme retention of the linear peptide, I suspect that the ammonium ions present in the mobile phase are acting as an ion pair, effectively shielding the negative charge and increasing the compound's retention.
To confirm that the DMSO was not in fact influencing the accessibility of the peptide solution to the mobile phase and the pH modifiers, I dissolved the peptide mixture in 5% aq. acetonitrile containing 0.1% ammonium hydroxide. I wanted to ensure the peptides were fully deprotonated prior to injecting onto the SNAP Ultra C18 cartridge, Figure 4.
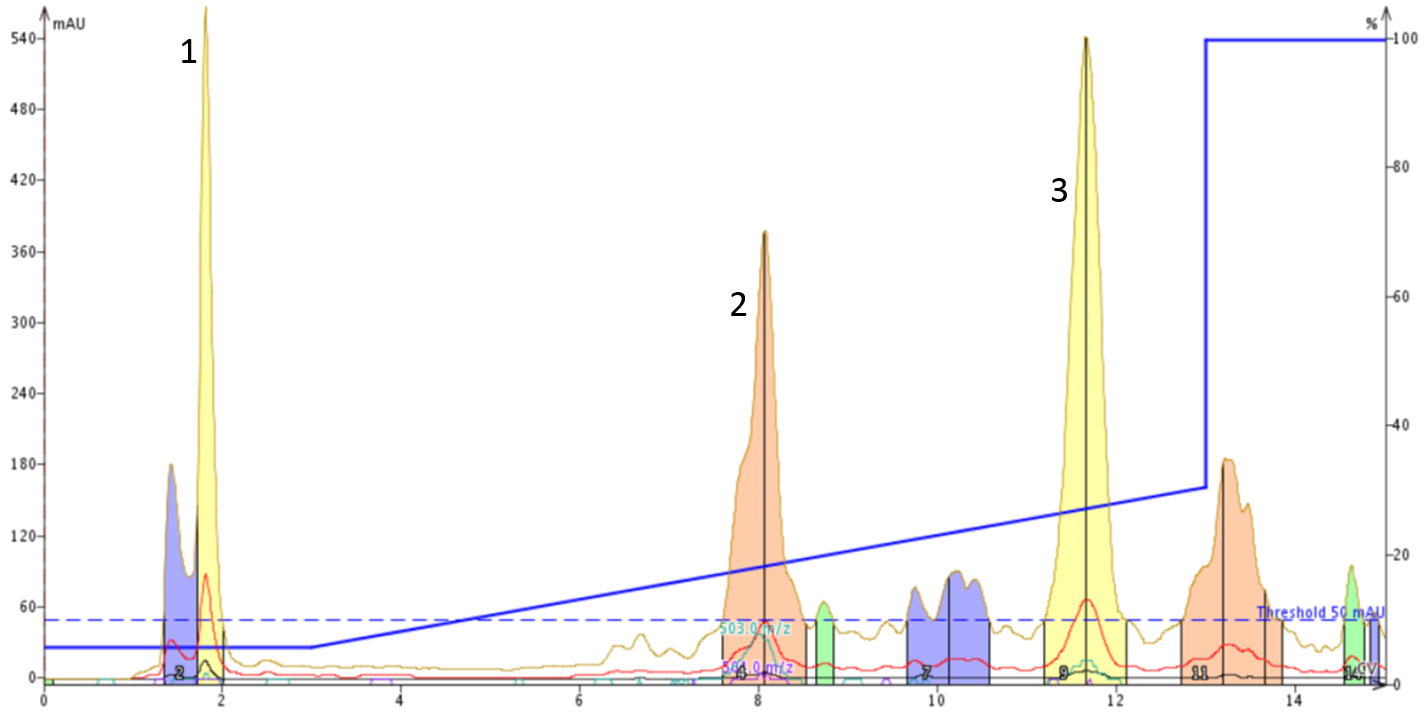
Figure 4: Optimized flash column chromatography gradient to separate linear from cyclized oxytocin dissolved in 5% aqueous acetonitrile containing 0.1% ammonium hydroxide. The gradient runs from 5% to 30% acetonitrile with mobile phase solvents containing 0.1% ammonium hydroxide.
Gratifyingly, the general peak separation pattern is maintained when the peptide mixture is dissolved in the initial mobile phase conditions. I should mention that the 20 mg crude sample was not fully solubilized in the 4-mL liquid solution prior to injecting onto the SNAP Ultra C18 cartridge. This is the likely cause for some of the observed peak shape decomposition. Interestingly, the cyclized peptide is not retained by the the column in the absence of DMSO. However, the linear peptide is still clearly resolved into two peaks, peaks 2 and 3 respectively. As the peptide mixture is dissolved in the basic mobile phase, I suspect that the two peaks arise from the partial deprotonation of the Tyr hydroxyl group (pKa ~10.46). The additional negative charge on such a small peptide would absolutely influence it’s retention by the C18 column, thereby yielding the observed peak distribution.
Although the resolution of flash column chromatography is less superior than HPLC, I have demonstrated here that with a little thought, a peptide can be purified from it’s contaminants by modulating the pH of the mobile phase. I will certainly be trying this technique for longer peptides in the future just to avoid the HPLC!
Interested in learning more about how flash chromatography can reduce your purification time and efforts? Follow the link below to find an application note.

