This application note describes optimization for a fully automated synthesis and on-resin cyclization of oxytocin enabled by Branches™ , a unique software feature of the Biotage® Initiator+ Alstra™ peptide synthesizer. The optimized protocol is then applied to the synthesis of isotopically labelled oxytocin. Also identified during this process is a strategy for an efficient separation of oxidized from reduced oxytocin via flash chromatography by modulating the pH of the mobile phase.
Introduction
Peptides continue to grow as an active area of research due to their ability to drive specific and selective interactions with a target. More importantly though, is their potential to interact with novel, previously considered undruggable targets. Cyclic peptides are particularly interesting due to increased serum stability, proteolytic resistance, cell permeability and potential for oral bioavailability.1–3
Peptide cyclization reactions are commonly performed in solution, requiring high dilution conditions to avoid intermolecular dimerization. On-resin strategies are attractive as immobilized peptides experience a pseudo-dilution environment, limiting intermolecular side reactions while favouring the desired intramolecular covalent bond. Methods that simplify automating the elongation and cyclization chemistries are very desirable.
Herein we present an optimized synthesis and on-resin cyclization strategy for oxytocin using Branches™ , a unique software feature that allows a skilled peptide chemist to visualize and specifically program on-resin synthesis of cyclic or branched peptides, Figure 1. The optimized synthetic protocol is then demonstrated through the synthesis of oxytocin containing an isotopically labeled leucine, where success is absolutely critical due to the high amino acid cost. Finally, a strategy to separate oxidized oxytocin from reduced oxytocin using flash chromatography is described.
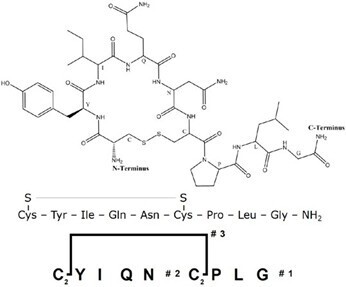
Figure 1. Sequence of oxytocin, including Branches™ representation of oxytocin.
Experimental
Materials
All materials were obtained from commercial suppliers and used without further purification; Sigma-Aldrich (diisopropylcarbodiimide (DIC), LC-MS grade water, LC-MS grade acetonitrile, triisopropyl silane (TIS), formic acid, ammonium in methanol (2 M)), Fisher Scientific (Fmoc-amino acids, Oxyma Pure, trifluoroacetic acid (TFA), 4-methylpiperidine, diethylether, N-chlorosuccinimide (NCS), hydrogen peroxide (50% aqueous)), Reagents, Inc. (dimethylformamide (DMF), acetonitrile, dimethylsulfoxide (DMSO), dichloromethane (DCM) and PCAS BioMatrix (Rink Amide ChemMatrix® resin).
Nα-9-fluorenylmethoxycarbonyl (Fmoc) amino acids contained the following side chain protecting groups: tert-butyl (Asp, Tyr), 4-methoxytrityl (Mmt, for Cys), and trityl (Gln, Asn).
Peptide synthesis
Peptides were synthesized using Fmoc-based solid phase peptide synthesis on a Biotage® Initiator+ Alstra™ automated microwave assisted peptide synthesizer. The synthesis was carried out using Rink Amide ChemMatrix® resin in either 10 mL or 30 mL reactor vials. Fmoc removal proceeded at room temperature twice, first for 3 minutes, then again for 10 minutes using 4-methylpiperidine in DMF (1:4). Coupling reactions proceeded with 4 eq. Fmoc-amino acid, 4 eq. DIC and 4 eq.
Oxyma Pure at 75 °C for 5 minutes for all residues except Cys, which was coupled at 50 °C to prevent side chain racemization. Upon synthesis completion, the resin was washed with DCM (x3) then diethyl ether (x3), repeated twice, then thoroughly dried under vacuum. The peptide was cleaved from the resin with TFA·H2O·TIS (95:2.5:2.5) for 2 hours at room temperature. The cleaved peptide was concentrated with a Biotage® V-10 Evaporation system, quickly rinsed with cold diethyl ether and lyophilized or immediately evaluated by analytical HPLC-MS. Crude peptides were analysed using a Biotage® Resolux 120 Å C18 column (5 µm, 150 x 4.6 mm) with an Agilent 1260 Infinity series HPLC coupled to an ESI-MS (AB Sciex 4000 QTrap) running Analyst software. The following solvent system was used: solvent A, water containing 0.1% formic acid and solvent B, acetonitrile containing 0.1% formic acid. The column was eluted using a linear gradient of 10–50% over 20 minutes. Peak integration and purity determination was conducted using MultiQuant 2.0.
Peptide purification
Peptides were purified using a Biotage® Isolera™ Dalton equipped with a 12 gram SNAP Ultra C18 cartridge. The peptide was dissolved in DMSO prior to injection onto the cartridge. The purification was conducted with the following mobile phase solvents:
- Solvent A, water + 0.1% TFA
- Solvent B, acetonitrile + 0.1% TFA
- Solvent C, water + 0.1% ammonium hydroxide
- Solvent D, acetonitrile + 0.1% ammonium hydroxide
For acidic purifications, an optimized linear gradient of 10–30% solvent B in solvent A was used to elute peptides from the cartridge. For basic purifications, an optimized linear gradient of 5–30% solvent D in solvent C was used to elute peptides from the cartridge. Fractionation was triggered by UV absorbance and monitored with mass detection for peak identification.
Positive ionization mode was used for purifications in acidic mobile phase (504 for cyclic peptide, 505 for linear peptide). Negative ionization mode was used for purifications in basic mobile phase (504 for cyclic peptide, 503 for linear peptide).
Results and discussion
Solid-phase peptide synthesis
The linear peptide was first synthesized with DIC/Oxyma activation and single couplings. Cysteine residues were coupled at 50 °C to limit side chain racemization. This approach yielded crude peptide with >95% purity and in 86% yield. Due to these results, the peptide elongation strategy was not optimized further. Linear oxytocin was then synthesized on a 0.5 mmol scale for rapid optimization of the Mmt removal and Cys oxidation steps.
Optimization of cysteine oxidation
Successful synthesis of the linear peptide was confirmed via microcleavage and the resin was subsequently divided for the Mmt removal and Cys oxidation chemistry evaluations.
Approximately 50 mg of dried peptidyl resin was transferred to a new reactor vial. For optimization of the Cys oxidation step, the Mmt removal was performed manually for two reasons:
- The steps required will inform the automated process.
- It ensured any incomplete oxidation was not due to incomplete Mmt removal.
Prior work has shown that a solution containing ammonia and hydrogen peroxide (2 eq. and 1.2 eq. respectively) can be used to promote Cys oxidation.4 This strategy was not effective for oxytocin, even after doubling the stoichiometric equivalents, Table 1. The observed inefficiency is presumably due to decreased conformation flexibility when compared to the mupain-1 example (4 vs. 9 intervening amino acids).
An alternative method demonstrated high yields for on-resin oxidation of oxytocin.5, 6 This strategy, utilizing N-chloroscuccinimide (NCS) as the oxidizing agent, was optimized further for this synthesis, Table 1. The optimized Cys oxidation requires 1 eq. NCS and only 5 minutes when heated to 50 °C for complete oxidation and minimal intermolecular dimer formed (<5% total peptide content), Figure 2.
Optimization of Mmt deprotection
There are many procedures published describing the removal of an Mmt protecting group. However, these procedures are often executed manually and yield varying degrees of success when automated. For this optimization, the reaction time and number of volume additions were evaluated for a variety of synthetic scales using freshly prepared TFA solutions, Table 2. During the optimization process, each Mmt removal step was monitored for completion by manual inspection of the reaction vessel. When the yellow solution colour no longer persisted, the deprotection was deemed complete. The optimized conditions for an average, 0.1 mmol scale synthesis required 4 additions, each reacting for 20 minutes.
|
Conditions |
Equivalents |
Temperature (°C) |
Time (min) |
Completion (%) |
|
NH3/H2O2 |
2/1.2 |
r.t. |
30 |
50 |
|
NH3/H2O2 |
4/2.4 |
r.t. |
30 |
50 |
|
NCS |
2 |
r.t. |
15 |
100 |
|
NCS |
4 |
r.t. |
5 |
100 |
|
NCS |
2 |
50 |
5 |
100 |
|
NCS |
1 |
50 |
5 |
100 |
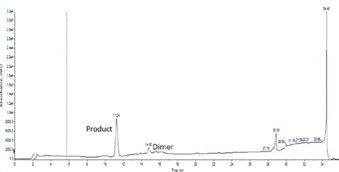
Figure 2. Crude analytical chromatogram for oxytocin cyclized under optimal Cys oxidation conditions using NCS. The desired product is contained in the main peak, with minimal intermolecular dimer present.
|
Resin (mg) |
TFA in DCM + 5% TIS |
Volume (mL) |
Time (min) |
Reactions |
|
25 |
1% |
2 |
10 |
7 |
|
50 |
1% |
5 |
20 |
4 |
|
300 |
1% |
9 |
20 |
10 |
|
50 |
2% |
2 |
20 |
2 |
|
400 |
2% |
5 |
20 |
4 |
|
1000 |
2% |
9 |
30 |
3 |
Applying optimized synthesis and oxidation conditions for preparation of isotopically labelled oxytocin
Isotopically labelled amino acids dramatically increases the cost of synthesis ultimately demanding high yield, and high crude purity. To demonstrate the utility of the optimized synthesis protocol described above, oxytocin containing an isotopically labelled leucine (Figure 3) was prepared.
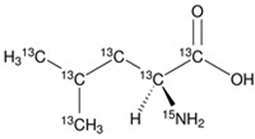
Figure 3. Isotopically labelled leucine indicating placement of heavy carbon and nitrogen isotopes.
Using the Branches™ software I readily assigned the optimized Mmt removal and Cys oxidation chemistry for the disulphide bond chemistry (Figure 1, step 3). The crude analytical HPLC, confirms the peptide was successfully synthesized and cyclized, Figure
4. Importantly, using the optimized conditions very little intermolecular dimer is observed in the analytical HPLC. The optimized conditions included standard DIC/Oxyma-mediated peptide elongation, Mmt removal after 4 additions of 2% TFA in DCM + 5% TIS each reacting for 20 minutes, and finally Cys oxidation with 1 eq NCS in DMF reacting at 50 °C for 5 minutes, Scheme 1.
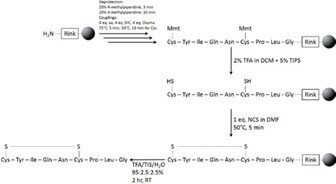
Scheme 1. Optimized, fully automated synthesis and oxidation of isotopically labelled oxytocin.
Cyclic peptide purification
Throughout the synthesis optimization steps, there were several opportunities to purify samples containing both oxidized and reduced oxytocin. Purification by flash chromatography was selected due to the high loading capacity of the flash cartridges, significantly reducing the time required to purify each synthesis. Despite many gradient optimization attempts, it was not possible to fully resolve the two peptides with standard mobile phase solvents containing acidic modifiers, Figure 5.
Oxidized and reduced oxytocin differ only by the presence or absence of a disulphide bond, which can be chromatographically exploited by adjusting the mobile phase pH to approximately 10, where the Cys side chain is fully deprotonated. Using this strategy baseline resolution of the two compounds was readily achieved, Figure 6.
Interestingly, the reduced peptide [M-2H]2- = 503 is retained by the stationary phase to a greater extent than the oxidized peptide [M-2H]2- = 504. This is likely due to an ion pairing interaction between the ammonium and sulfonium ions, increasing the overall hydrophobicity of the peptide when compared to the minimal hydrophobicity gains from the presence of a disulphide bond.

Figure 4. Crude analytical chromatogram of oxytocin synthesized with isotopically label Leu and the optimized synthetic conditions. Peak overlap with purified cyclic oxytocin indicates that the sample is indeed fully cyclized as no shift in retention time is expected for peptides differing in isotopic abundance (left). Peak integration of the TIC at this retention time shows molecular weights for the peptide differing by 7 Da, as expected for the isotopically heavy peptide (right).
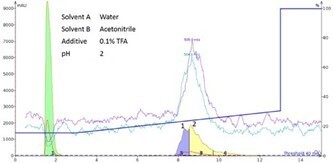
Figure 5. Flash chromatogram resulting from the separation of reduced and oxidized oxytocin using acidic mobile phase. Linear([M+2H]2+ = 505) (Peak 1) and cyclic ([M+2H]2+ = 504) (Peak 2) peptides are not resolved.
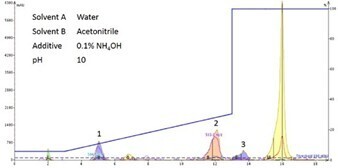
Figure 6. Flash chromatogram resulting from the separation of reduced and oxidized oxytocin using basic mobile phase. Linear ([M-2H]2- = 503) (Peak 2 and 3) and cyclic ([M-2H]2- = 504) (peak 1) peptide are baseline resolved.
Conclusion
Herein we present an optimized, fully automated synthesis and on-resin cyclization followed by rapid purification of oxytocin. High yielding crude syntheses of the linear peptide was then subdivided for optimization of both the Cys oxidation and the Mmt removal steps. The use of Branches™ , a unique software feature on the Biotage® Initiator+ Alstra™ simplified programming the orthogonal chemistry required for Mmt removal and disulphide bond formation. Separating linear from cyclized oxytocin proved challenging by flash column chromatography with standard RP mobile phase solvents, but was successfully achieved by increasing the pH above the pKa of the Cys side chain sulphur atoms.
References
1. Bockus, A. T.; McEwen, C. M., Lokey, R. S., Cur. Top. Med. Chem. 2013, 13, 821–836.
2. Davies, J. S., J. Pep. Sci. 2003, 9, 471–501.
3. Wang, C. K.; Craik, D., J. Pep. Sci. 2016, 106, 9011–909.
4. Biotage® application note AN093: Automated Synthesis of Cyclic Peptides on Biotage® Initiator+ Alstra™ .
5. Postma, T. M.; Albericio, F. E., J. Org. Chem. 2014, 17, 3519–3530.
6. Postma, T. M.; Albericio, F., Org. Let. 2013, 15, 616–619.
Literature Number: AN115
