Introduction
Large numbers of related peptides are often required for many types of research, for example in proteomic studies of post-translational modifications. While this sometimes can be achieved by split-and-mix combinatorial synthesis, parallel synthesis of large numbers of peptides offers distinct advantages. These include that there is no need for decoding, well- characterized compounds can be used, and that compounds can easily be transferred to other platforms, for example microarrays for parallel screening.
Glycosylation is one of the most abundant post-translational modifications of proteins as well as peptides, and aberrant glycosylation is a hallmark of malignant transformation.1 Mucin-type O-glycosylation is particularly important in relation to cancer-associated autoantibodies, because cancer cells generally produce short truncated O-glycans such as Tn (GalNAcα1-O-Ser/Thr), T (Galβ1-3GalNAcα1-O-Ser/Thr), and STn (NeuAcα2-6GalNAcα1-O-Ser/Thr).2 Thus, there is a great need for chemically and chemo-enzymatically synthesized well-defined glycopeptides, for example, for the construction of glycopeptide microarrays.
The research groups of Blixt and Jensen3,4 have described a high-throughput O-glycopeptide discovery platform for seromic profiling, using the Syro II parallel peptide synthesizer for high-throughput glycopeptide synthesis where over 1700 O-glycopeptides were synthesized (see Figure 1).5
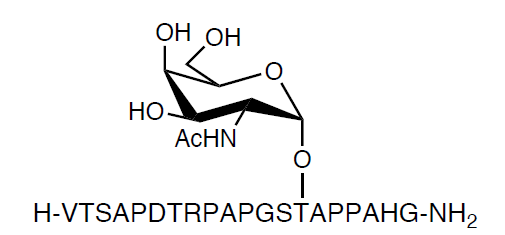
Figure 1: Example of 20-mer O-glycopeptide Tn-Muc1a
Experimental
Materials
All materials were obtained from commercial suppliers; Sigma-Aldrich (acetonitrile, formic acid and triethylsilane (TES)), Iris Biotech GmbH (Fmoc-amino acids, DMF, NMP, N-[(1H-benzotriazol-1-yl)(dimethylamino)methylene]- N-methylmethanaminium hexafluorophosphate N-oxide (HBTU), 1-hydroxybenzotriazole (HOBt), trifluoroacetic acid (TFA), piperidine and N,N-diisopropylethylamine (DIPEA)) and Rapp Polymere GmbH (TentaGel S Rink Amide resin). Milli-Q (Millipore) water was used for LC-MS analysis.
Nα-9-fluorenylmethoxycarbonyl (Fmoc) amino acids contained the following side-chain protecting groups: tert-butyl (Ser, Thr), 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl (Pbf, for Arg), tert-butoxycarbonyl (Boc, for Lys) and trityl (Trt, for Asn, Gln, His). The glycosylated amino acid (Fmoc-Thr(Ac4-β-Glc)-OH was synthesized using glycosylation chemistry as described in the literature,6 and Fmoc-Thr(Ac3-α-GalNAc)-OH was purchased from Sussex Research (Canada).
Synthesis and analysis
General solid-phase synthesis of peptides and glycopeptides
Peptides and glycopeptides were prepared on a Syro II parallel peptide synthesizer (Biotage AB, Sweden) by standard SPPS methods on TentaGel S Rink Amide resin (loading 0.24 mmol/g) on a 2.5 μmol scale in 2 mL reactor vials using 2 x 48 position reactor blocks. Nα–Fmoc deprotection was performed using piperidine-DMF (2:3) for 3 min followed by piperidine-DMF (1:4) for 12 min and then washing with NMP (x4).
Nα-Fmoc amino acids (4.0 eq.) were coupled using HBTU (3.8 eq.), HOBt (4.0 eq.) and DIPEA (8.0 eq.) in DMF for 120 min and then washing with NMP (x4). Capping of the unreacted N-terminal amines was performed following each coupling step using two consecutive treatments with acetic anhydride-DCM solution (1:4) of 15 min each and thorough washing with NMP (x4) was undertaken in between treatments. The peptides and glycopeptides were released from the solid support, and all acid-labile protecting groups were removed by treatment with TFA/TES/H2O (95:2:3) for 2 h. The TFA solutions were concentrated by nitrogen flow and the compounds were precipitated with cold Et2O and crude materials were collected as white pellets after centrifugation. MALDI-TOF MS was performed on representative samples (Figure 2).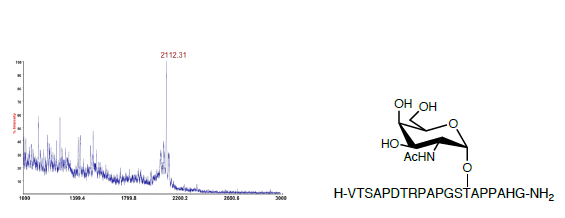
Figure 2. MALDI-TOF crude product
Deacetylation of glycopeptides
Prior to deacetylation, the crude glycopeptides were mixed with Stratosphere PL-HCO3 MP SPE resin in order to remove the excess of ion-paired TFA as follows: typically, a 96-member batch of crude glycopeptide pellets (1–4 mg each) were taken up in 500 µL each of water-MeOH (4:1) solution and loaded into a 96-well filter plate (one glycopeptide per well) contains Stratosphere PL-HCO3 MP SPE resin (2.0 g, evenly distributed in the 96 wells) pre-conditioned in MeOH. After 5 min incubation the filter plate was drained under vacuum and thereby the filtrate from each well collected separately in a 2 mL/well, 96-well receiving plate.
The Stratosphere PL-HCO3 MP SPE resins in the filter plate were then washed with 100 µL fresh MeOH per well and likewise collected in the receiving plate under vacuum suction. MeOH was removed from the receiving plate where- upon the remaining water phase was lyophilized. The base-free solid crude glycopeptides were then dissolved in 300 µL of MeOH and the pH adjusted to 9 (as judged from pre-wetted pH strips) with a 0.015M solution of MeONa in MeOH. After 10 h of incubation at rt, 250 µL of NaOAc 0.1M solution pH 6 was added in order to quench the basicity and thereby giving rise to glycopeptide stock solutions. The stock solutions were stored at -20 °C until required for on-chip glycopeptide capture microarray printing.
Results & discussion
An overview of the synthesis of over 1700 O-glycopeptides, which were needed for the construction of novel glycopeptide microarrays is shown in Figure 3. The N-terminal amino- functionalized glycopeptides and peptides were printed on a microarray slide to provide glycopeptide microarrays. By introducing a capping step during chemical solid-phase glycopeptide synthesis, selective enrichment of N-terminal glycopeptide end products were achieved on an amine-reactive hydrogel coated microarray glass surface. This allowed for high- throughput display of large numbers of glycopeptides.
Several libraries of a total of over 1700 glycopeptides and peptides were prepared. Each peptide and glycopeptide was validated by mass spectrometry. This was combined with subsequent on-slide enzymatic glycosylation. As a proof-of-concept it was demonstrated that MUC1 glycopeptides could be assembled and detect autoantibodies in vaccine induced disease-free breast cancer patients and in patients with confirmed disease at time of diagnosis.
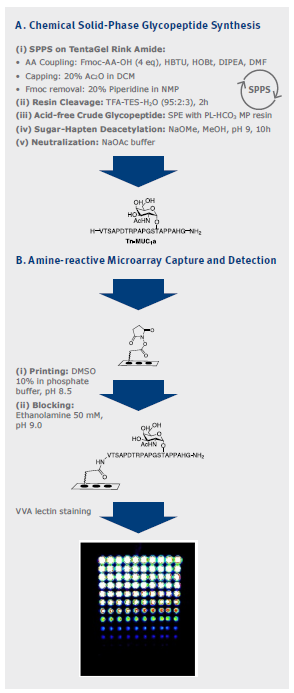
Figure 3: Overview of the parallel synthesis of glycopeptides, their release and deprotection, and printing to form glycopeptide microarrays.
Conclusion
More than 1700 glycopeptides and peptides were successfully prepared on a Syro II parallel peptide synthesizer in a high- throughput manner. This provided multiwell low scale synthesis of peptides and GalNAc-glycopeptides combined with direct printing without prior purification for a facile and economical approach to display hundreds of glycopeptides. These glycopeptides and peptides were used for the construction of a novel glycopeptide microarray. Biomarker microarrays are becoming valuable tools for serological screening of disease-associated autoantibodies. For further details on the development of the synthetic screening microarray platform for facile display of post-translationally O-glycosylated peptides, please see the publications by Blixt, Jensen and co-workers.3-5
References
- Hakomori, S.; Proc. Natl. Acad. Sci. USA 2002, 99, 10231–10235.
- Brockhausen, I.; Biochim Biophys Acta 1999, 1473, 67–95.
- a) Kracun, S.; Cló, E.; Clausen, H.; Levery, S. B.; Jensen, K. J.; Blixt, O.; J. Proteome Res., 2010, 9, 6705–6714.
b) Steentoft, C.; Schjoldager, K. T.; Cló, E.; Mandel, U.; Levery, S. B.; Pedersen, J. W.; Blixt, O.; Jensen, K. J.; Clausen, H.; Glycoconjugate J., 2010, 6, 571–582. - Cló, E.; Kracun, S; Nudelman, A. S; Jensen, K. J.; Liljeqvist, J.; Olofsson, S.; Bergström, T.; Blixt, O.; J. Virol., 2012, 86, 6268–6278.
- Blixt, O.; Cló, E.; Nudelman, A. S.; Sorensen, K. K.; Clausen, T.; Wandall, H. H.; Livingston, P. O.; Clausen, H.; Jensen, K. J.; J. Proteome Res. 2010, 9, 5250–5261.
- Yao, N.; Fung, G.; Malekan, H.; Ye, L.; Kurth, M. J.; Lam, K. S.
Carbohydr. Res. 2010, 345, 2277–2281.
Acknowledgements
Prof. Ola Blixt and co-workers, Prof. Knud J. Jensen and Dr. Kasper Kildegaard Sørensen and co-workers at the University of Copenhagen.
Literature number: AN096
