Jan 23, 2023 11:23:43 AM
By Bob Bickler

slash column chromatography has been practiced by chemists since the 1970s. That practice requires the silica in the column be properly wetted to remove trapped gasses and ensure uniform flow (remember those days of not letting air into the column?). Today, with automated flash chromatography systems and pre-packed columns as the norm, chemists ask me – do I really need to pre-equilibrate my column?
In this post I explore the impact on chromatography that equilibration, and lack thereof, has on separation performance.
In all my years as a chemist who specializes in chromatography this is the one topic that generates the most discussion. I have met with chemists who always equilibrate before loading their crude material and those that never equilibrate. Which is correct? The answer is – it depends. It depends on your specific purification goals and the retention of your product of interest.
Let me explain by detailing what happens when you pump solvent through a dry silica bed
- Silica, which is dry and active (adsorptive), quickly absorbs the solvent into its pores and at the same time adsorbs it as well onto the silica surface. This is especially true of solvents with some polarity.
- The absorption process displaces trapped air from the silica’s pores. Full air displacement is accomplished in two to three column volumes.
- The adsorption process generates heat through a process known as the heat of adsorption. The temperature increase can be quite dramatic to the point where solvents can boil within the column (higher flow rates and more polar solvents increase the amount of heat generated). The “heat band” that is created dissipates quickly as it rides with the solvent front and exits the column within one column volume. However, I have seen where the temperature increase is so great that the frits melt creating extremely high back-pressure, especially with a polar solvent content of greater than 10%.
- After the initial heat band passes, the silica surface continues to adsorb the polar solvent component. If the equilibration volume is sufficient, the amount of adsorbed polar modifier, e.g. ethyl acetate, achieves a steady-state having the same concentration at the top of the cartridge as the bottom.
OK, now that we understand what happens during equilibration, think about what happens when a sample is purified on a new cartridge without equilibrating it first
- Air displacement – will this cause a detection issue? Not normally and your compounds are unlikely to elute before one column volume anyway.
- Solvent and sample adsorption generates heat. What are the issues with this?
- Pressure increases (the hotter the temperature generated the higher the pressure). As mentioned above the amount of pressure can be high enough to over-pressurize your flash system or even burst your column! This can be a safety issue. Also, depending on your sample component's concentration, an increase in pressure may cause in-column sample precipitation, not beneficial.
- Chromatographic adsorption/desorption kinetics are altered. This can lead to undesired separation issues such as unresolved compounds and low purity fractions.
- If your compounds are even semi-volatile, when the solvents heat up you run the risk of reducing your purified compound yield because they may begin transitioning from a dissolved solid/oil to a vapor and exit the cartridge into the atmosphere.
Opining further, altering of the chromatography can be catastrophic, especially if you load all of your reaction mixture and end up with a poor separation generating very low purity fractions.
In an effort to show the impact of equilibration on purification, I created an experiment where a 5-component mix is separated on a 10-gram Biotage® SNAP Ultra column both with and without equilibration.
The linear gradient used for both was a 5% ethyl acetate to 40% ethyl acetate in hexanes over 10 column volumes (CV). The load was 100 mg (0.1 mL) and the gradient flow rate was 36 mL/min; new, unused columns were used for each test. Detection was UV photodiode array 200-400 nm.
Two nearly identical methods were created but one included the equilibration step. Equilibration was performed with 5% ethyl acetate at 100 mL/min for 3 CV (51 mL) prior to sample load.
The results from the equilibrated cartridge show five well-resolved peaks, Figure 1.
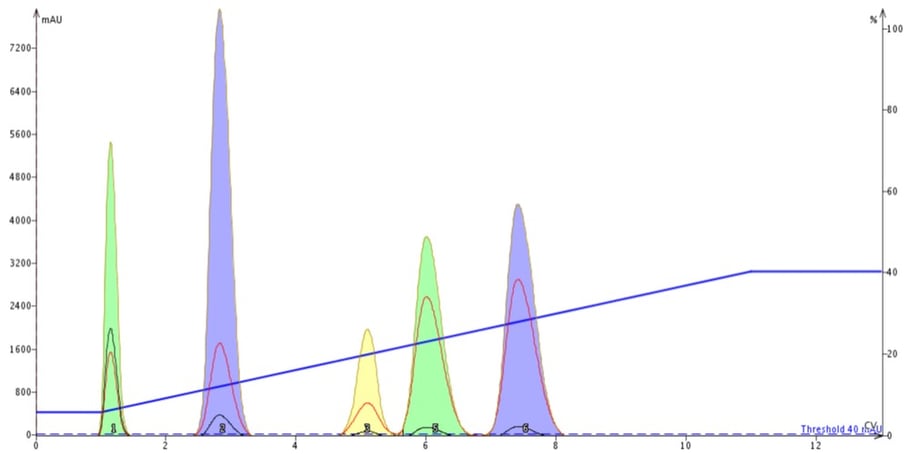 Figure 1. When a flash column is equilibrated its separation performance is predictable and reproducible.
Figure 1. When a flash column is equilibrated its separation performance is predictable and reproducible.
The results from the non-equilibrated cartridge, however, show severe separation degradation. The first two peaks have merged and the last three peaks have broadened, which has impacted their resolution, Figure 2.
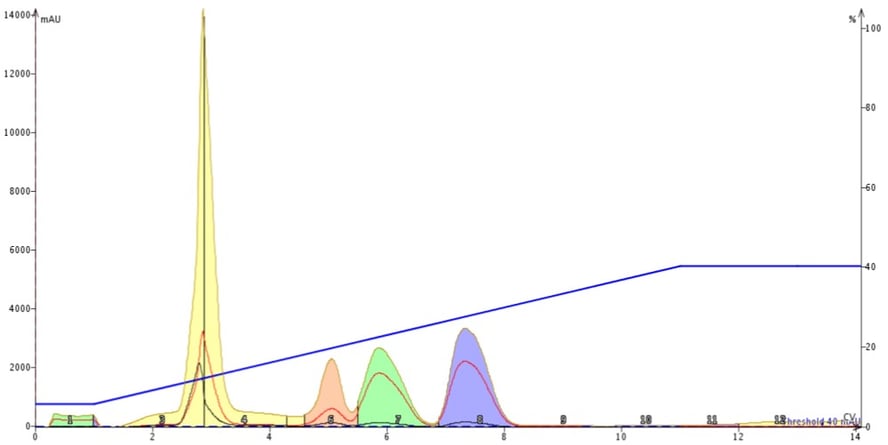
Figure 2. An example of the impact not equilibrating a silica column has on chromatographic performance. The low, broad green peak around 1 CV is vaporized solvent which contains some compounds. The yellow peak contains the first two eluting compounds while the last three peaks have broadened diminishing the separation between them.
So, why the difference? Well, as mentioned above when dry, active silica comes in contact with solvent, the solvent gets hot. Heat interferes with compound adsorption so those compounds that are most lipophilic (Rf values > 0.5) are most susceptible to altered chromatography. This can be seen in the data below where the five compounds' Rf values range from 0.9 to 0.15, Table below.
|
Peak |
Rf |
|
1 |
0.90 |
|
2 |
0.63 |
|
3 |
0.50 |
|
4 |
0.31 |
|
5 |
0.15 |
The last three eluting peaks with Rf values 0.5 and lower are retained and separated, though with reduced resolution compared to an equilibrated cartridge. This data suggests that lack of equilibration is not all that detrimental to good chromatography if your compounds are retained on TLC with Rf values <0.5. Remember, your best separations and loading capacity are created when your compound(s) of interest have TLC Rf values between 0.1 and 0.4.
To me, however, I would rather sacrifice a little time and solvent to achieve the best possible separation than end up with a potentially sub-optimal purification of my valuable product.
The choice ultimately is yours. If your target product is retained (Rf <0.5) you can try not equilibrating but be aware that there are some potential downsides.
Do you equilibrate your flash cartridges before starting the gradient?
For more flash chromatography information, download our white paper - Successful Flash Chromatography

{kind=link}