Jan 30, 2023, 5:11:21 PM
By Elizabeth Denton

One common technique in HPLC for improving difficult peptide separations is to extend the column length, a topic I explored for flash chromatography in a previous post. However, alternative purification strategies are sometimes necessary as the purification bottleneck grows with increasing peptide library size, both in number and scale.
In this post, I explore using two identical size cartridges in series with each packed with a different stationary phase. I wanted to try this to see if I could improve peptide purity with the ultimate goal of reducing the time demand of peptide purification.
So this probably sounds weird to most of you. It’s definitely not normal to connect two HPLC columns, packed with different stationary phases, inline for a single purification. But that’s exactly what I’m going to explore.
There is some precedent for this type of thing though. Two dimensional HPLC is often used for differential fractionation in proteomics experiments. The first dimension is often an exchange type chromatography (strong cation or anion) which is then followed by standard reversed phase chromatography and mass spectrometry analysis.
While the two dimensions I’m working with aren’t truly orthogonal, there is potentially sufficient selectivity differences between C18 and C4 alkyl chains to motivate a hypothesis that this will work as well.
I decided to use the Biotage® SNAP Bio line of cartridges because they are available with the two different functionalities bonded to the same underlying silica (same particle and pore size).
Before I could get started with the fun part, I first needed to evaluate the purification efficiency of the C4 cartridge alone using my crude 18A sample (see previous post for crude and SNAP Bio C18 purified analytical data). Based on my experience last time, I decided to inject 300 µL of the crude peptide in DMSO onto the cartridge using my standard 20% to 70% gradient and monitoring for the m+2 (1101), m+3 (734.4) and m+4 (551) ions using the Biotage® Isolera Dalton™ 2000, Figure 1.
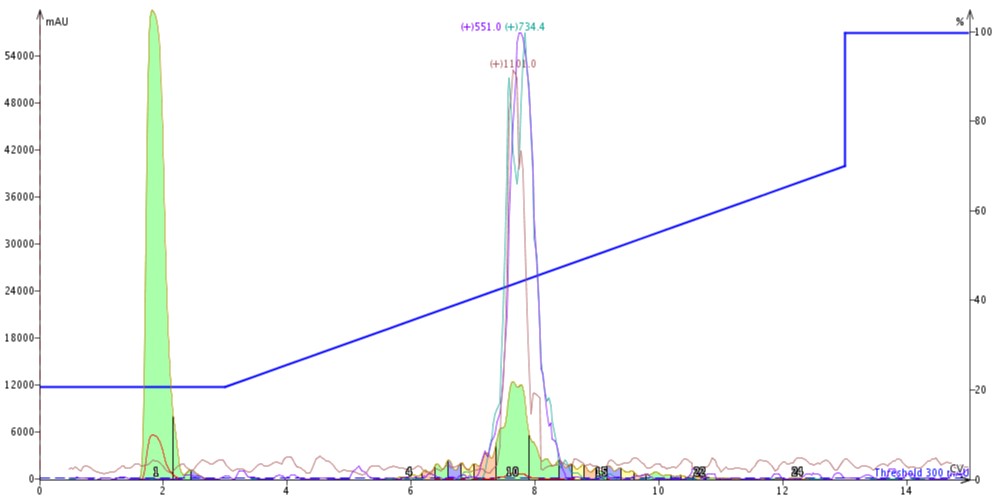
Figure 1: Crude 18A purified using a SNAP Bio C4 cartridge. The peptide is eluted from the column with a gradient running from 20% to 70% acetonitrile. The m+2 (red), m+3 (green) and m+4 (purple) masses are monitored for peak identification.
The peak shape is certainly not gaussian, suggesting that there may be some residual impurities co-eluting with the desired peptide product. The C4 did resolve a leading impurity not separated using the C18 media. And as expected, the peptide is retained slightly less by the C4 alkyl chains than by the C18 alkyl chains (~7.5 CV elution vs ~8.5 CV for C18). I combined and concentrated fractions 10 and 11 (green fractions) for analytical HPLC which confirmed my suspicions that the main peptide has measurable impurities, Figure 2.
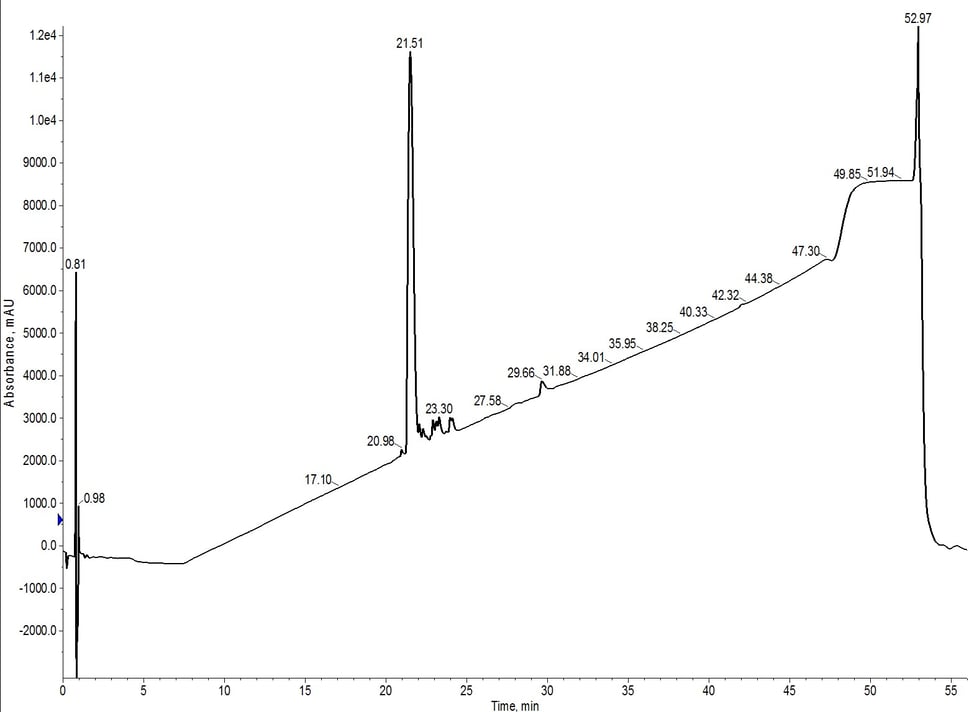
Figure 2: Analytical HPLC evaluation of 18A purified using a single C4 cartridge (fractions 10 + 11). While moderately successful, the purification was not as efficient as that observed previously for a single C18 SNAP Bio cartridge.
The flash chromatography did remove a significant fraction of the impurities, but further purification may be required to move forward in biological assays (depending on the assay constraints). Although it's not a true direct comparison due to the lower loading amounts, the C18 cartridge alone using the same gradient, generated a more pure final product. The crude analytical HPLC of 18A shows a significant number of impurities with greater hydrophobicity than the desired peptide. When purified using the C4 cartridge, the lower carbon content and resulting less lipophilic environment likely allowed for co-elution of these impurities.
With this information in hand, I connected the C4 cartridge to the top of a C18 cartridge (C4-C18), loaded 300 µL crude 18A in DMSO and ran the exact same gradient, Figure 3. I kept the flow rate constant here as well so that any purification efficiency comparisons would not be muddied by differing linear velocities. Connecting two cartridges in line though does increase the back pressure by a factor of two.
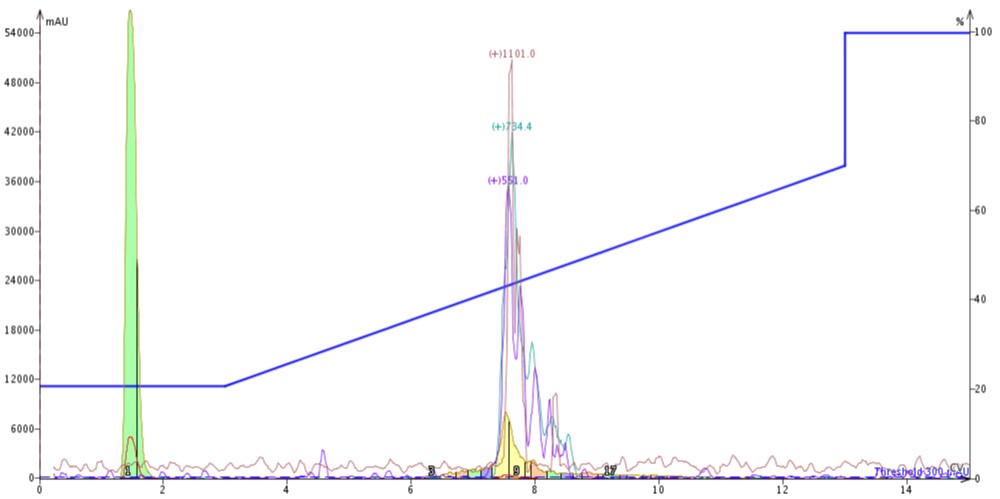
Figure 3: Purification of crude 18A completed by connecting a 10 gram SNAP Bio C4 cartridge to the top of a 10 gram SNAP Bio C18 cartridge.
The peak shape, despite doubling the column length, actually sharpens when compared to the C4 or C18 cartridge alone (likely due to increased backpressure in the system). Chromatographically, the peptide now elutes at about 7.5 CV, suggesting that the C4 cartridge is dominating the elution profile. After combining and concentrating fractions 8 and 9 (yellow fractions), the analytical HPLC shows an increase in the overall sample purity when compared to the C4 purification, Figure 4.
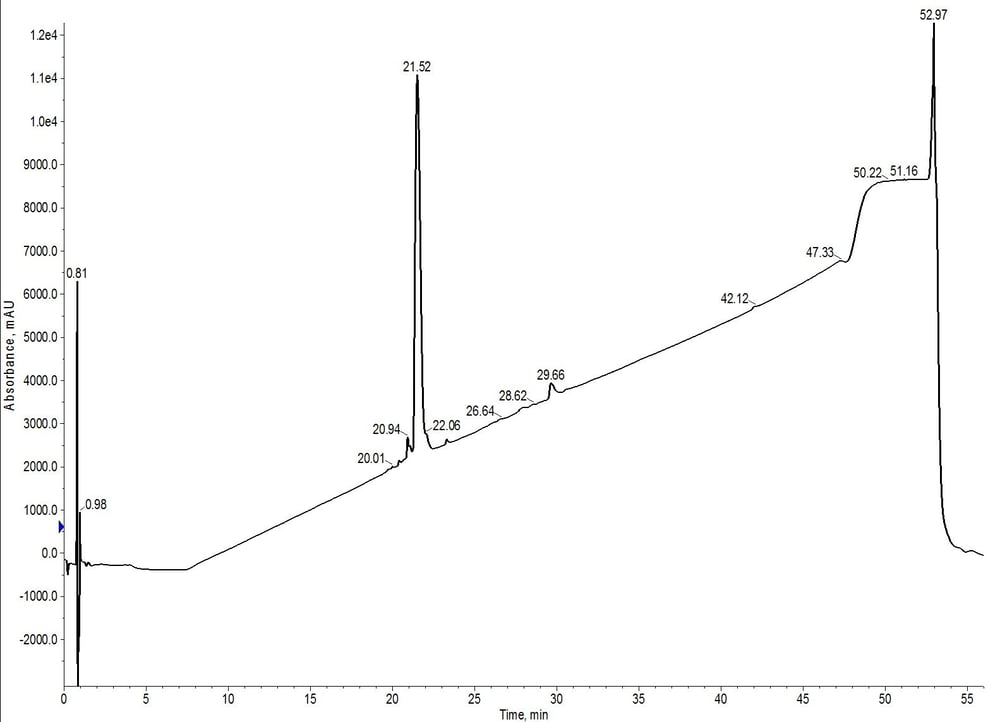
Figure 4: Analytical HPLC of 18A purified using C4-C18 inline cartridges. The hydrophobic impurities have been removed when compared to the C4 cartridge alone. However, a relatively significant hydrophilic impurity remains at 20.94 minutes.
For the next injection, I switched the cartridge order. With the C18 cartridge positioned on top of the C4 cartridge (C18-C4), I injected 300 µL of crude 18A in DMSO and again used the same gradient as above, Figure 5.
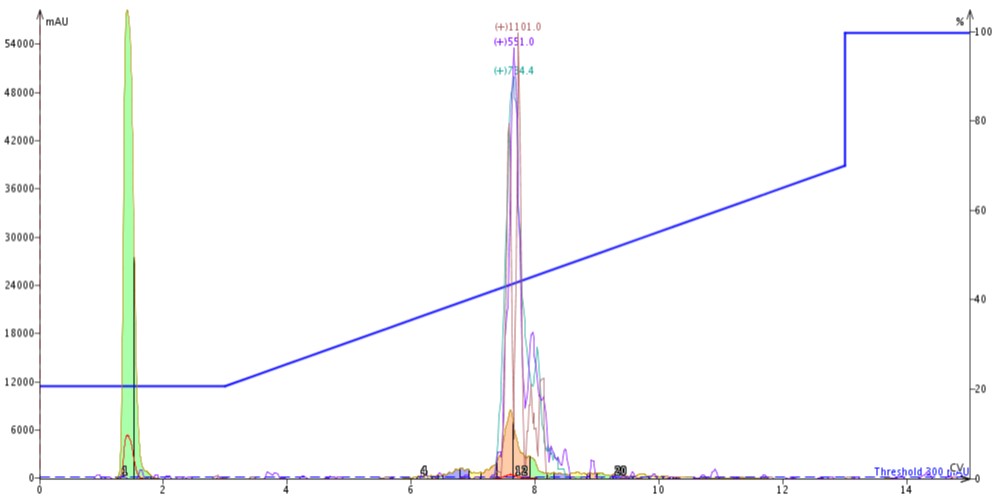
Figure 5: Purification of crude 18A completed by connecting a 10 gram SNAP Bio C18 cartridge to the top of a 10 gram SNAP Bio C4 cartridge.
The peak shape is slightly degraded when compared to the C4-C18 separation, but the overall elution profile remains the same (approximately 7.5 CV elution). The trailing shoulder (only resolved using the C18 media) is equivalently resolved from the main product peak, but the leading impurity is now significantly resolved from the main peptide. This is clearly demonstrated in the analytical HPLC of fractions 11 and 12 (pink fractions) where the leading impurity has been reduced, Figure 6.
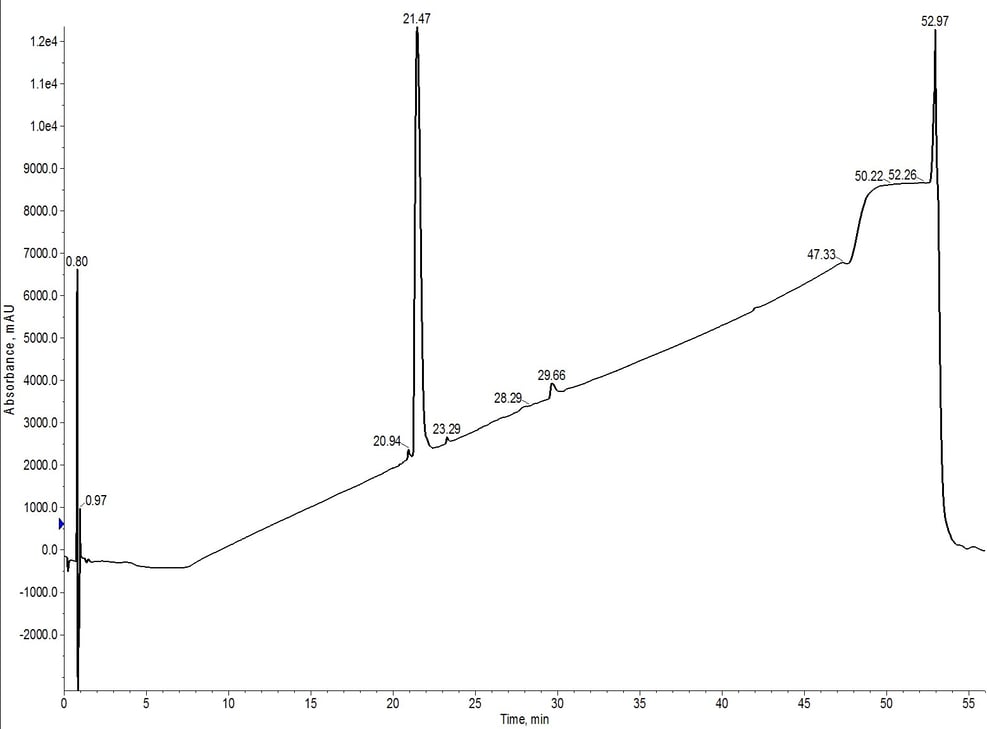
Figure 6: Analytical HPLC of 18A purified using C18-C4 inline cartridges. The hydrophobic impurities have been removed when compared to the C4 cartridge alone and the leading impurity has also been significantly reduced.
So what is going on here? Let’s take a closer look at the SNAP Bio C18 and SNAP Bio C4 purifications alone, Figure 7. There are two important features to point out. In the purification mediated by C4 alkyl chains, there is a clear impurity peak eluting prior to the product peaks (blue arrows), which is not nearly as well resolved in the C18 mediated purification. Alternatively, the trailing shoulder (orange arrows) is much more resolved with the C18 mediated purification than the C4 mediated purification. These differences are due to the differences in selectivity exhibited by the C4 and C18 stationary phases.
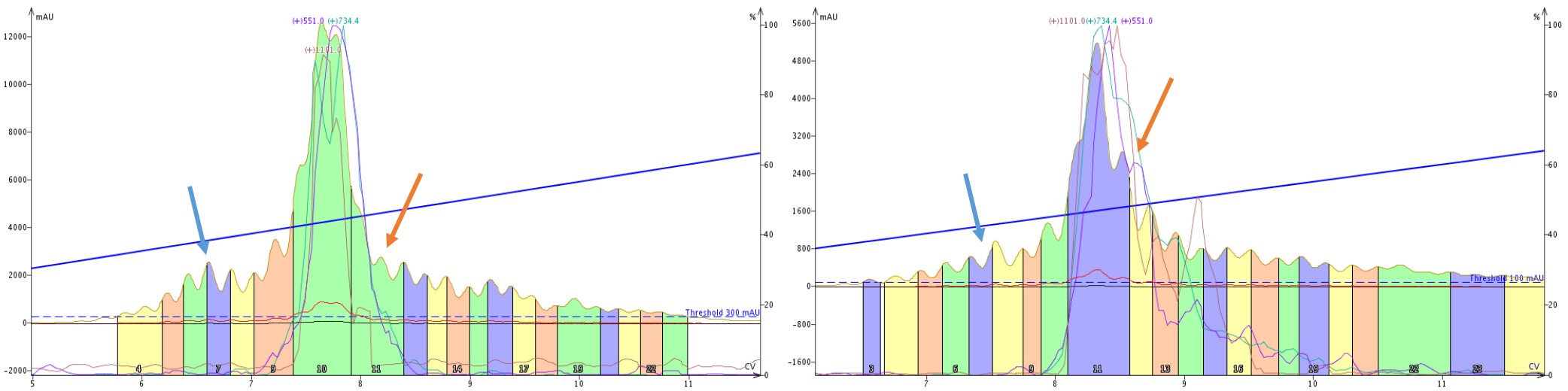
Figure 7: Side by side comparison of the elution profile when purifying crude 18A with a single SNAP Bio C4 (left) or SNAP Bio C18 (right). A leading impurity (blue arrow) is significantly more resolved with the C4 cartridge than the C18, whereas a trailing peak (orange arrow) is resolved with the C18 cartridge but not the C4 cartridge.
When we take a closer look at the tandem purifications, those same features immediately are apparent but differ depending on the order in which the cartridges are mounted, Figure 8. The leading impurity (blue arrows) is well resolved only when the C18 cartridge is encountered first by the sample. Interestingly though, the trailing shoulder (orange arrows) seems to maintain resolution regardless of cartridge order.
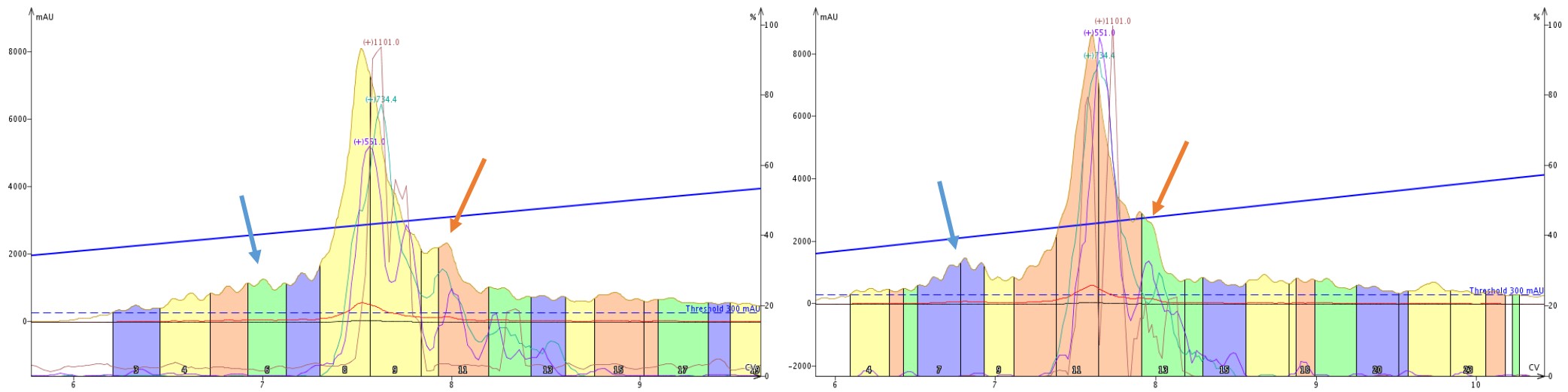
Figure 8: Side by side comparison of the elution profile when purifying crude 18A with a SNAP Bio C4 then C18 cartridge (left) or SNAP Bio C18 then C4 cartridge (right). A leading impurity (blue arrows) is significantly more resolved with the C18 cartridge placed first, whereas a trailing peak resolution appears to be unaffected by cartridge order.
The selectivity difference can be calculated by measuring the elution volume of each peak of interest (leading, product, or trailing peaks) and the void volume, determining the capacity factor (k’), and calculating the selectivity factor (α), Table 1.
|
Retention volume (mL) |
||||
|
C18 alone |
C4 alone |
C18-C4 |
C4-C18 |
|
|
lead peak |
99 |
204 |
||
|
product peak |
125 |
114.5 |
228 |
225.5 |
|
trailing peak |
134 |
237 |
239 |
|
|
void volume |
26 |
27.5 |
42.5 |
44 |
|
Capacity factor (k') = (Vc-V0)/V0 |
||||
|
lead peak |
2.60 |
3.80 |
||
|
product peak |
3.81 |
3.16 |
4.36 |
4.13 |
|
trailing peak |
4.15 |
4.58 |
4.43 |
|
|
Selectivity (α) = k'2/k'1 |
||||
|
Product peak/lead peak |
1.22 |
1.15 |
||
|
Trailing peak/product peak |
1.09 |
1.05 |
1.07 |
These values clearly demonstrate the selectivity differences for C4 vs C18 when comparing the leading peak with the product peak. The hydrophobicity of the C18 stationary phase causes the impurity to be more significantly retained, decreasing the separation between the leading impurity from the desired product. When the C18 cartridge is placed first in series, the selectivity loss is initially felt, but the presence of the C4 cartridge in line restores the selectivity and ultimate separation. If the C18 cartridge is second in series, any selectivity gains achieved by the C4 cartridge is eliminated.
This was a cool experiment! Running the cartridges in line resulted in a more pure compound with fewer injections, ultimately requiring less purification time. It would be interesting to expand this evaluation to other peptides, particularly altering the hydrophobic content to more thoroughly evaluate if this pattern holds. My guess is that it won’t, but this could still be a very useful method for purifying difficult compounds quickly and efficiently with flash chromatography.
If you would like to learn more about using flash chromatography to purify your peptides, click the link below.
