Jan 31, 2023, 3:55:43 PM
By Elizabeth Denton

More and more groups are exploring the utility of peptides with an ever widening variety of applications. And although peptides are getting cheaper to purchase outright, many groups are continuing to bring peptide synthesis in house. As more groups join the peptide community, I frequently encounter questions about the basics of peptide synthesis.
In today's post, I'd like to cover a little history of solid phase synthesis as well as highlight some differences between the chemistries.
Chemistries and reagents for amide condensation reactions have existed for decades and been utilized to great effect by organic chemists globally. As interest in bioactive peptides grew during the mid 20th century though and along with it the need for synthetic analogues, it quickly became apparent that the "standard" solution phase strategies would not be sufficient.
Solution phase peptide synthesis is typically very arduous and laborious - requiring long coupling reaction times and a need for recrystallization or column chromatography between each amino acid coupling. After making several analogues of bradykinin (9 amino acids), Bruce Merrifield sought an alternative and began developing methods for solid phase peptide synthesis, efforts for which he later won the Nobel Prize in Chemistry. The beauty of Merrifield's strategy is that chemical reagents can be reacted with a reactive moiety on a solid support, then removed by a simple filtration step, improving the throughput of peptide synthesis.
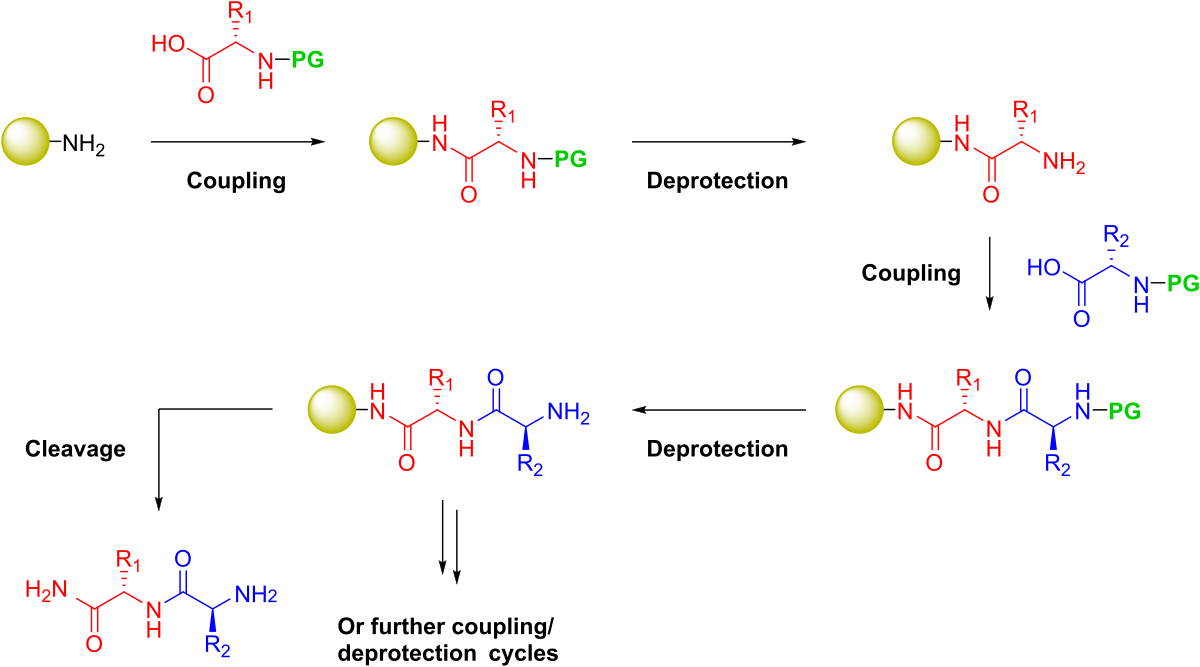
Figure 1. General steps required to synthesize a peptide on solid phase. Protecting groups (PG) will be present on the amine nitrogen for iterative deprotection as well as any potentially reactive side chain groups.
To implement a filtration-based strategy, Merrifield combined a functionalized, insoluble polymeric support and protecting group chemistry, enabling the iterative addition of each amino acid building block, Figure 1. Two types of protecting groups were required for this to be successful: 1) an amine-reactive reversible protecting group that could be liberated from the N-terminus and 2) protecting groups for the various polar and charged side chain moities that would be resistant to the frequent N-terminal deprotection conditions, yet sensitive to the conditions which liberate the final peptide sequence from the solid support.
With these conditions in mind, Bruce worked to developed the strategy now known as the Boc synthesis strategy. In this strategy, the N-terminal amine is protected with a Boc group, Figure 2, which can then be removed with moderate concentrations (usually 25-50%) of trifluoracetic acid. Functional side chains are protected using chemical moities stable to the repeated TFA treatment, but can still be liberated during treatment with the resin cleavage cocktail. The final peptide is cleaved from the resin originally using hydrofluoric acid (HF). Other cleavage conditions have been identified due to the safety concerns associated with HF use. The principle in Boc chemistry is simple: protecting group and global cleavage from the solid support are removed based upon relative acid sensitivity with the amine nitrogen protecting group being the most sensitive (least acidic conditions) and global cleavage from the resin being the least sensitive (most acidic conditions).

Figure 2. Mechanism for removal of a Boc protecting group from the N-terminus of a growing peptide chain using trifluoracetic acid (TFA).
Several years after Merrifield's introduction of the Boc SPPS strategy and using chemistry pioneered by Louis Carpino, Atherton and Sheppard applied the Fmoc amine protecting group to solid phase peptide synthesis. Rather than take advantage of acidolysis sensitivity, Fmoc-based chemistry combines base sensitivity with acid sensitivity, Figure 3.
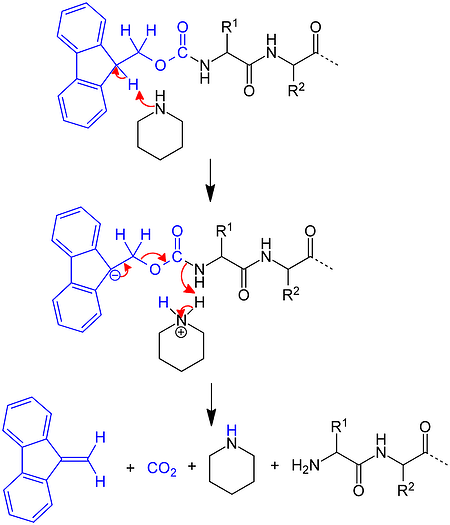
Figure 3. Mechanism for removal of an Fmoc protecting group from the N-terminus of a growing peptide chain using piperidine.
Fmoc-based SPPS has revolutionized synthetic accessibility to chemists in a number of ones. First, and possibly most importantly is that the chemical routinely employed in Fmoc chemistry exhibit a much greater safety profile than those used in Boc chemistry. Repeated handling of relatively concentrated TFA solutions and handling of HF has been eliminated. This improved safety profile has enabled inexperienced chemists to undertake peptide synthesis efforts with relative ease and success.
But secondly, exploited acid and base lability has welcomed the introduction of a huge diversity of side chain protecting groups. Introduction of new, orthogonal protection strategies enables on-resin chemistries and modifications that would be much more difficult or even impossible with traditional Boc protocols. This becomes even more relevant when we consider the complexity of peptides that are now routinely synthesized using Fmoc protocols.
Interested in performing peptide synthesis in your lab? Follow the link below to learn more about what you'll need to get started.
