Nov 2, 2023 2:00:00 PM
By Austin Schlirf

The dynamics of solid phase peptide synthesis at the resin surface makes it particularly difficult to create head to tail macrocycles. Dimerization, steric hinderance, and C-terminal access all reduce the efficiency of a standard coupling for head-to-tail cyclization. Here I discuss the effect that resin loading capacity plays on this hindered reaction.
In a previous blog, I showed that head-to-tail cyclization is possible on resin using Asp and Glu side-chained linked peptides. However, at most, a 28% crude purity was attained for the cyclic species. One of the significant impurities, dimerization, is directly caused by interactions between 2 adjacent peptide chains which makes up ~1/3 of the impurities found in the crude cyclized material. I first repeated my experiment using our Glu-linked peptide and a standard 0.49mmol/g Rink amide polystyrene resin to attempt to improve our linear product crude purity but got similar results. For cyclization, we used our standard double coupling method using DIC/Oxyma coupling reagents.
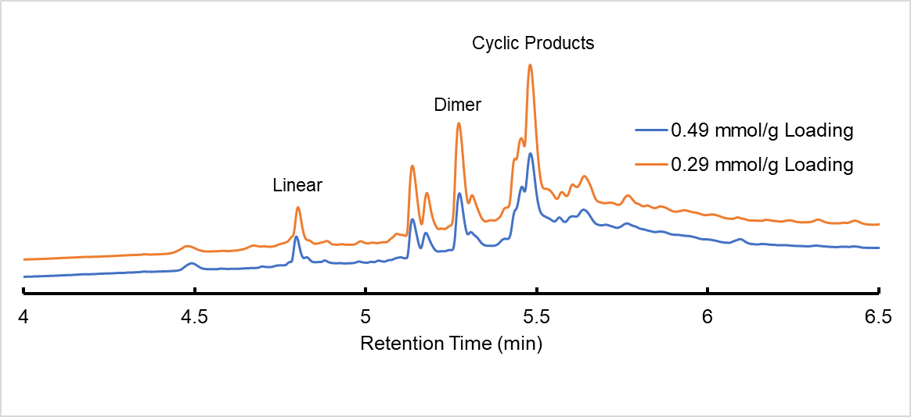
Figure 1. Stacked chromatogram of Glu linked 0.49mmol/g and 0.29mmol/g loading cyclic reaction product.
This synthesis was repeated for the 0.29 mmol/g loaded resin to compare the effect of resin loading capacity on crude purity as a strategy to reduce interchain interaction. Here my thinking is if I decrease the density of active sites then I may spread out my peptide enough to promote intrachain cyclization over dimerization. As you can see in Figure 1, the samples are quite similar each with ~30% of identifiable cyclic product in the crude which includes our dimer that has been converted into a cyclic compound. In these samples I only had ~3% left of linear product and ~12% linear dimer after the cyclization reaction.
| Compound | 0.29 mmol/g Loading | 0.49 mmol/g Loading |
| Linear Peptide | 3.45% | 2.90% |
| Unidentified Impurities | 8.59% | 10.36% |
| Dimeric, Linear Peptide | 13.15% | 11.90% |
| Cyclic Product(s) | 27.39% | 32.35% |
Table 1 comparison of products between 0.29 mmol/g and 0.49 mmol/g loading under the same synthesis conditions
For this experiment changing up my loading seems to have not made much of an improvement. Even still, I'm unable to distinguish between monomeric cyclic and dimeric cyclic compounds chromatographically as with this gradient they coelute. However, when comparing the amount of product, I get from cleaving the resins, I get back a higher mass return from my 0.49 loaded resin comparably to my 0.29 loaded resin; ~1.7x as one would expect based on the loading ratios.
If we think of the peptide in this experiment as a floppy noodle, then the N-terminus will have much more freedom than the C-terminus. Therefore, my -COOH group which is the active species can only react with free NH2 that come into its vicinity. At 0.29mmol/g loading there still seems to be too much active site density for dimerization to be neutralized i.e the N-terminus of my noodle finds the C-terminus of another. I could further reduce the loading on my resin however this would provide even less peptide per synthesis. I think next I will try to see if reducing my peptide length can improve the purity and reduce side reaction potential of my cyclic peptides.
What type of cyclic species are you seeing and what are you using to overcome them? If you want to read more on cyclic peptides, see the link below!

