Feb 2, 2023 3:10:33 PM
By Elizabeth Denton

Sometimes the most interesting peptides are also the absolute worst to work with. Whether it's synthetic difficulty, or solubility issues, or purification difficulty, or worst of them - all of the above - the experiments must go on! This is the case for amyloid beta and many of the amyloidogenic peptides currently being studied.
In today's post, I'll use this particularly difficult peptide to handle as a case study for some strategies to improve your purification efficiency.
The β-amyloid peptide family are particularly difficult....everywhere. They can be challenging to synthesize as they contain a large number of hydrophobic amino acids, many clustered in patches along the 40 or 42 amino acid lengths. However, handling these peptides after cleaving from the resin often causes greater challenges. With minimal solubility and a high propensity to aggregate or fibrillate in solution, these peptides are extremely difficult to purify as well.
I synthesized β-amyloid1-40 using my Biotage® Initiator+ Alstra™ peptide synthesizer and got pretty decent results considering the minimal effort I put into thinking about the synthesis protocol and handling during/after cleaving from the resin, Figure 1. 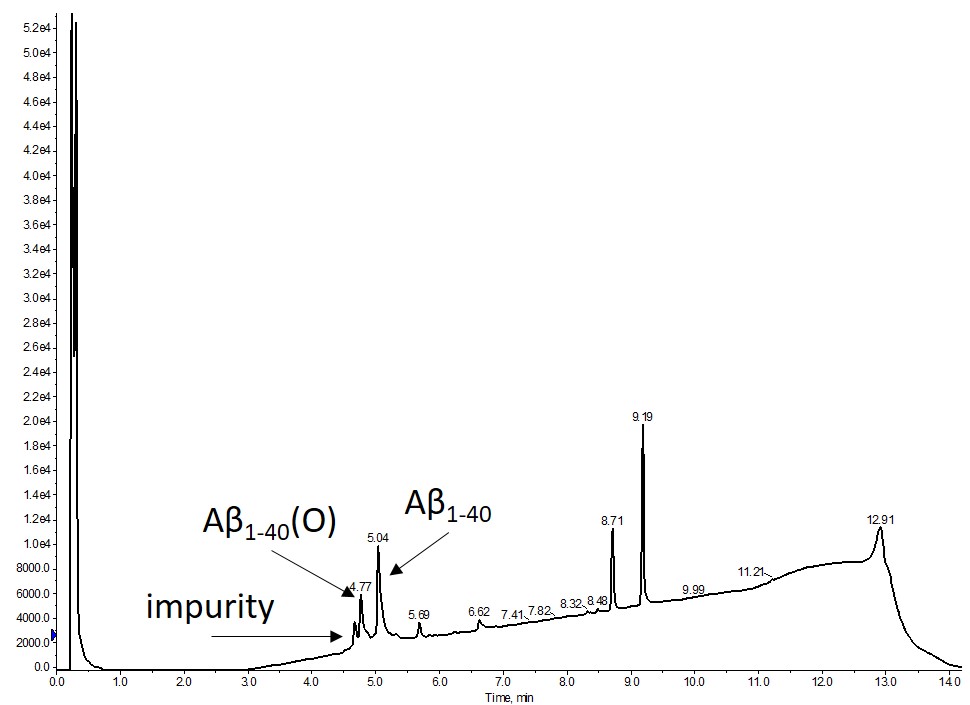
Figure 1. Crude analytical HPLC of synthetic A-β1-40 dissolved in DMSO. The major product is the desired with a significant fraction containing the oxidized peptide (O). It is important to note that no preventative steps were taken to minimize any Met oxidation from occurring during synthesis or cleavage from the resin.
Thankfully, some guidelines for re-solubilization of purified β-amyloid that yield monomeric peptides in solution have been published. Rather than try to reinvent the wheel, I decided to follow the reported guidelines to solubilize my crude peptide sample. For these experiments I wanted to evaluate two of the strategies - using DMSO and aqueous solutions at basic pH.
An important consideration is the mobile phase modifier. Fully monomeric and solubilized peptide does me no good for purification if it immediately aggregates once it hits the column. I did experiment a bit with TFA-modified solvents, as this is the most common system used for peptide purification, but my results were a little weird and inconsistent (data not shown). So given the recommendation to dissolve the peptide in basic aqueous solvents, I decided to switch my mobile phase modifier to 0.1% NH4OH for the actual experiments of interest.
I optimized the gradient a bit, starting with a focused gradient and ultimately settling on a step gradient to fully resolve each of the three major peaks present in the crude chromatogram. I then dissolved ~100 mg of the crude peptide in 500 µL of DMSO for purification using the step gradient, Figure 2.
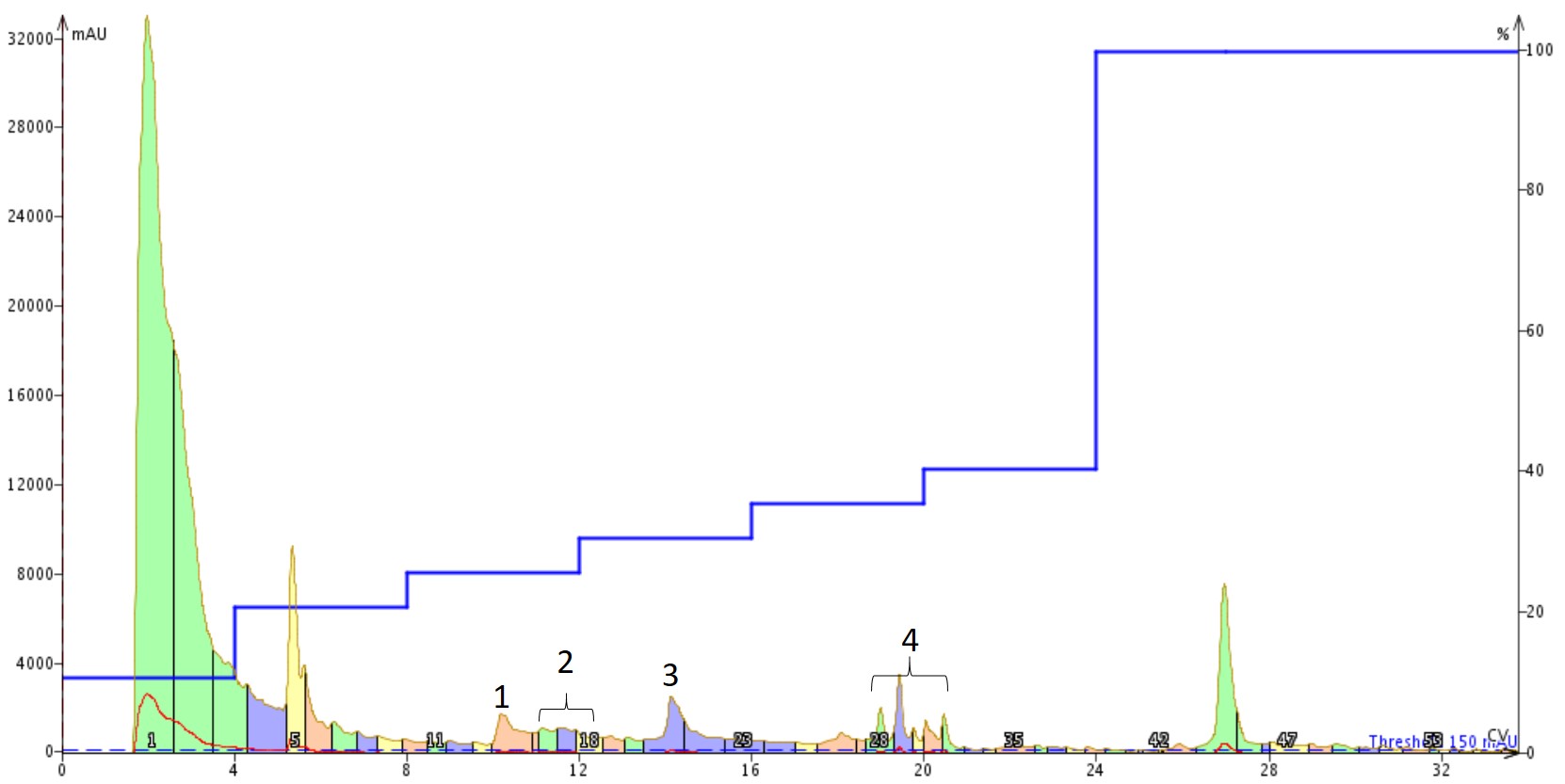 Figure 2. Purification of Aβ1-40 dissolved in DMSO using a Sfar Bio C18 column connected to an Isolera™ Dalton 2000 system with a somewhat optimized step gradient. The four labelled peaks were further characterized analytically for specific peptide content.
Figure 2. Purification of Aβ1-40 dissolved in DMSO using a Sfar Bio C18 column connected to an Isolera™ Dalton 2000 system with a somewhat optimized step gradient. The four labelled peaks were further characterized analytically for specific peptide content.
This chromatogram surprised me some, to be honest. The DMSO solution injected was completely transparent, suggesting that it was free from aggregates. However, the shape and number of peaks present suggests otherwise though. I decided to concentrate the fractions and investigate the relative purity of each of the labelled peaks, Figure 3.
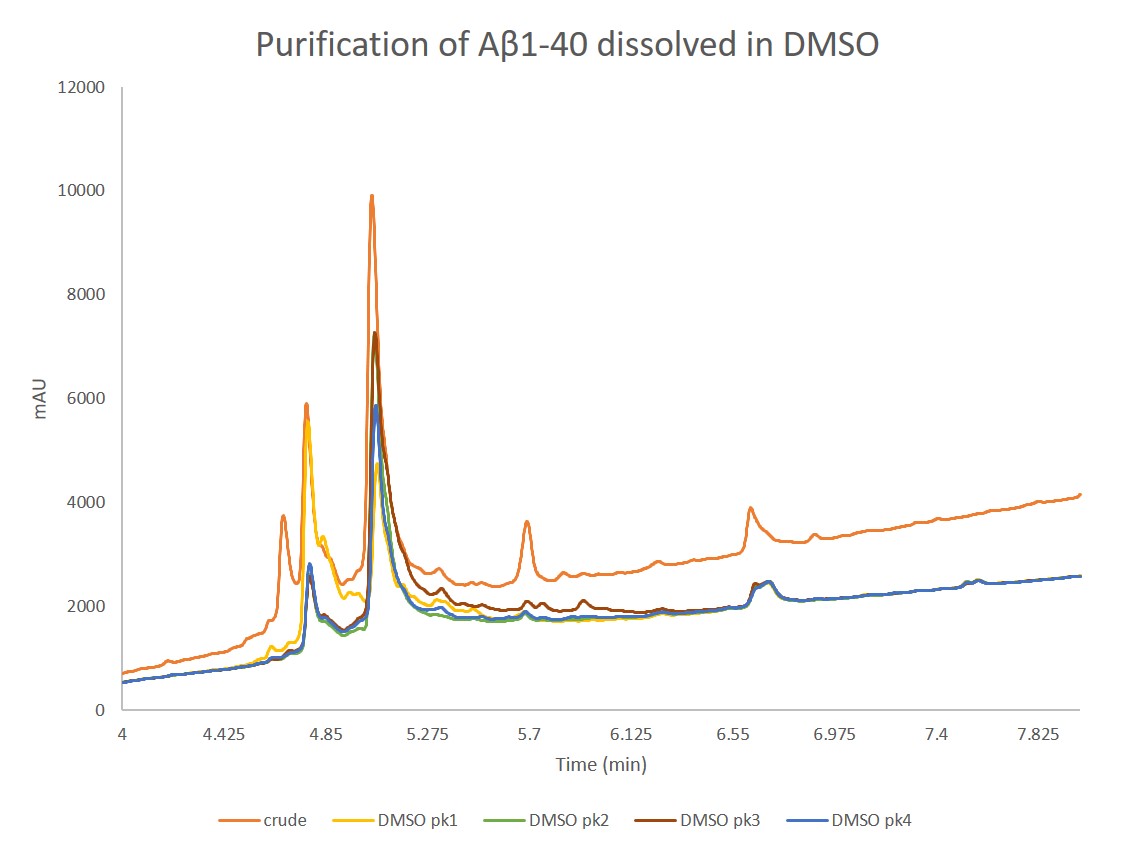 Figure 3. Analytical HPLC of concentrated samples collected from peak 1, peak 2, peak 3, and peak 4 overlaid with the crude peptide. Peak 1 contains predominantly the oxidized product, as expected, while the three other peaks contain decreasing fraction concentrations of the desired peptide. Interestingly, the analytical HPLC for peak 2 and peak 3 are nearly identical in composition.
Figure 3. Analytical HPLC of concentrated samples collected from peak 1, peak 2, peak 3, and peak 4 overlaid with the crude peptide. Peak 1 contains predominantly the oxidized product, as expected, while the three other peaks contain decreasing fraction concentrations of the desired peptide. Interestingly, the analytical HPLC for peak 2 and peak 3 are nearly identical in composition.
Interestingly, all fractions contain both the oxidized and the desired peptide, just with different relative concentrations. The presence of oxidized peptide may be the results of a couple different issues:
- poor resolution during the purification itself
- oxidation of the desired peptide during handling after purification was completed.
- aggregation of oxidized peptide with the desired peptide during the purification
For peaks 2 and 3 in particular, the relative ratios of oxidized peptide: desired peptide (ox:pdt) are nearly identical, with the most significant difference due to a broader product peak in the peak 3 sample. While subtle, this factor should not be overlooked. These two samples have very different chromatographic behaviour during the initial purification- particularly peak 2 - despite similar analytical compositions. The chromatographic differences suggest that most likely a conformation or oligomerization difference is occurring between these two samples. With the tools at my disposal today, I can't confirm what those conformational differences, may be unfortunately.
I decided for the second purification to include some acetonitrile as part of the dissolution solvent because I typically do not ether precipitate my crude peptides. As a result, the side chain protecting groups are still present in the dried solid and significantly reduce the total solution solubility in water. In this case though, the sample began to gel (early stage aggregation and possibly fibrillation) almost immediately. If we purify the same quantity of crude peptide, this time dissolved in 25% MeCN(aq) + 1% NH4OH, using the same step gradient we see entirely different chromatographic behaviour, Figure 4.
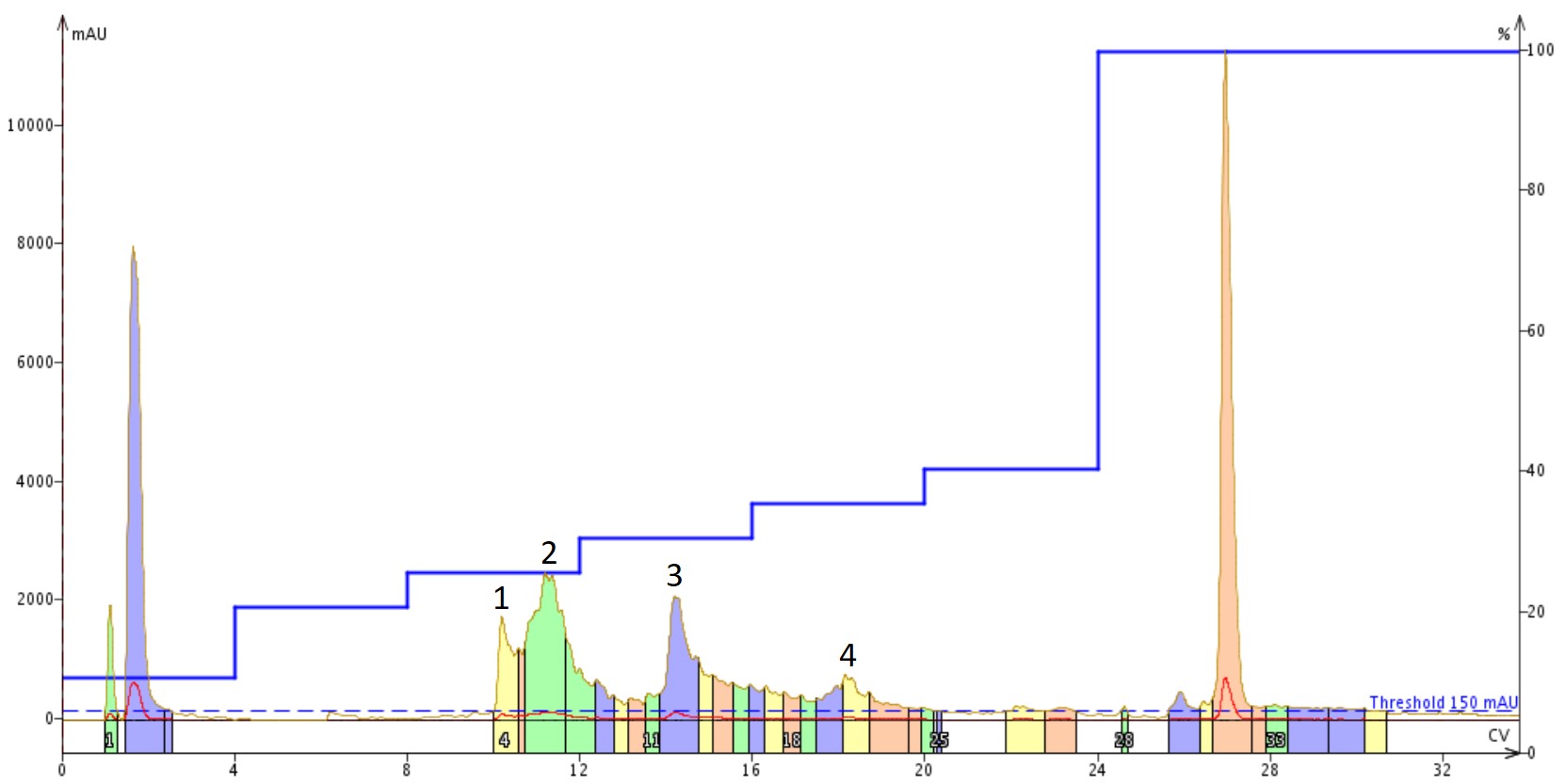
Figure 4. Purification of Aβ1-40 dissolved in a 25% MeCN(aq) +1% NH4OH solution using a Sfar Bio C18 column connected to an Isolera™ Dalton 2000 system with a somewhat optimized step gradient. The four labeled peaks were further characterized analytically for specific peptide content.
As in the previous injection, there are still more peaks than would have been anticipated from the original analytical. Given the evidence of immediate aggregation (gelling) though, this isn't too surprising. Importantly though, peak shape in general is much improved suggesting fewer conformational discrepancies present in the solution and likely improving the overall purification efficiency. The four labeled peaks were further analyzed analytically, Figure 5.
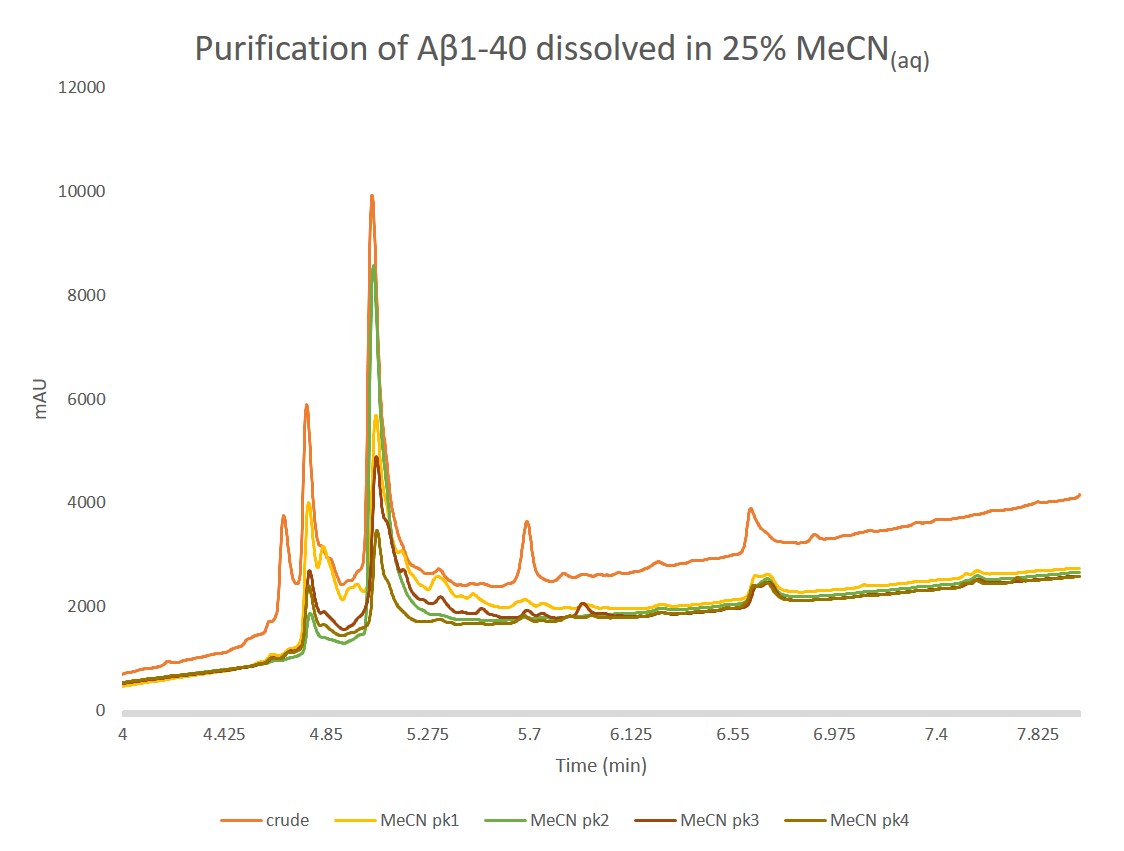
Figure 5. Analytical HPLC of concentrated samples collected from peak 1, peak 2, peak 3, and peak 4 overlaid with the crude peptide. Peak 1 contains a large fraction of the oxidized product, as expected. Importantly, peak 2 contains predominantly the desired peptide and only a small fraction of the oxidized product. The two remaining peaks also contain mixtures of oxidized and desired product, but with increasing relative concentrations. Given these increasing ratios and alternative chromatographic retentions, peaks 3 and 4 are likely differential oligomerization states of the desired peptide.
Good! This looks much better, and more in line with what I would have expected from the crude analytical chromatogram. Peak 1 contains a significant amount of both the oxidized and desired peptides, but we see that peak 2 is substantially the desired product, suggesting this is likely the monomeric, desired product. Based on these results, this set of solvation conditions certainly yielded a greater purification efficiency than the DMSO solvated purification.
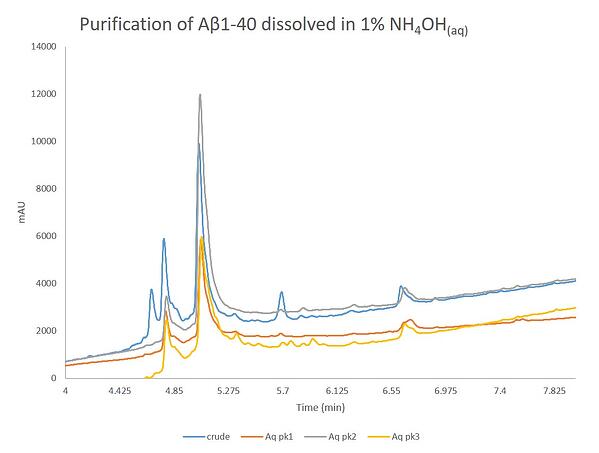
Finally, I decided to dissolve ~100 mg of the crude peptide sample in 1% NH4OH(aq) and then purified the crude material, Figure 6. These conditions, with relatively quick dilution into alternative buffers, have been demonstrated to be most efficient at not only breaking up any pre-formed aggregates, but also maintaining the monomeric state in solution.
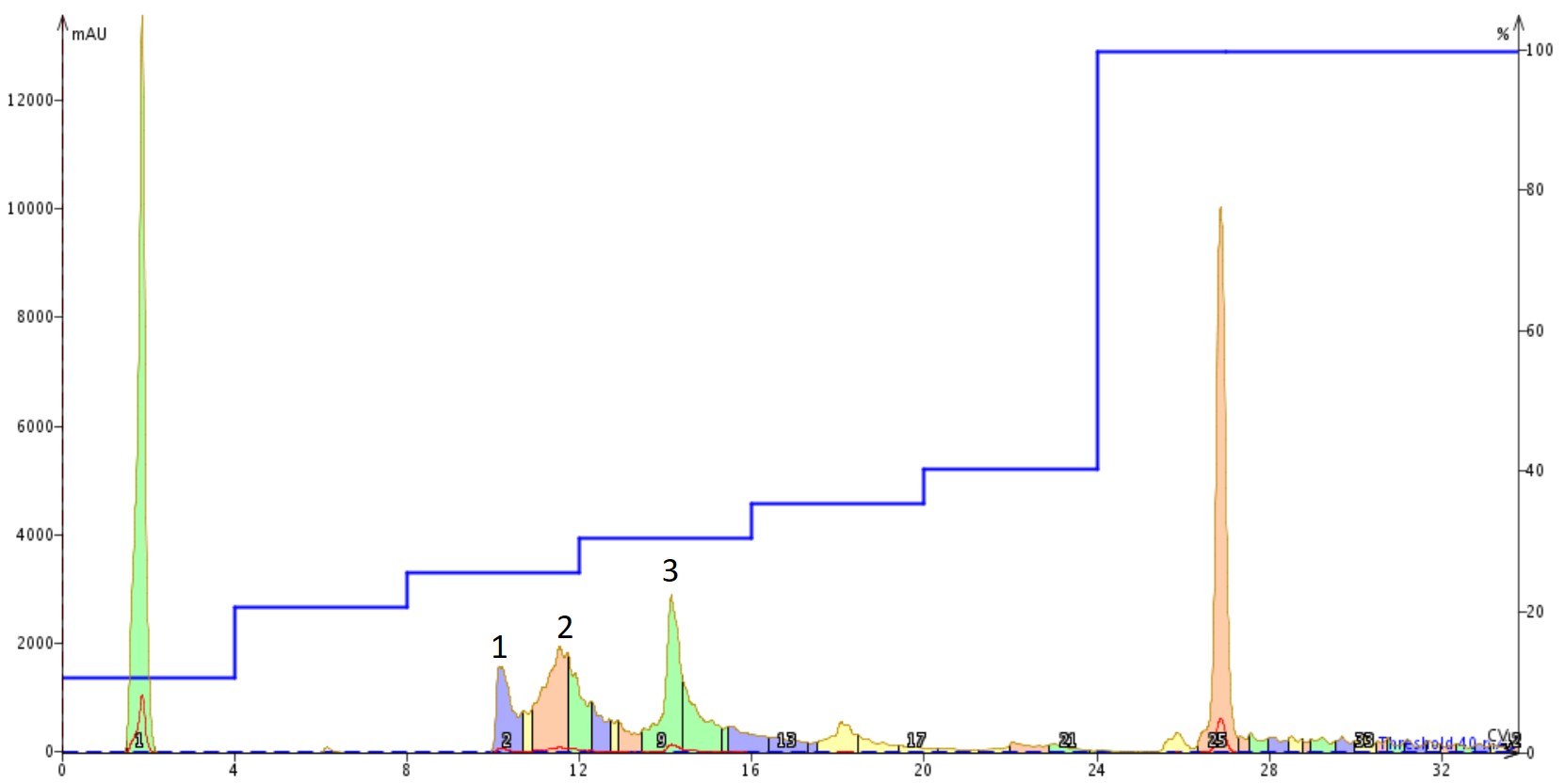
Figure 6. Purification of Aβ1-40 dissolved in a 1% NH4OH(aq) solution using a Sfar Bio C18 column connected to an Isolera™ Dalton 2000 system with a somewhat optimized step gradient. The three labeled peaks were further characterized analytically for specific peptide content.
This purification absolutely turned out the best. The peak shape and resolution is improved even over the previous purification effort. Although, there was a small amount of turbidity of the dissolved sample before the purification began, it doesn't appear to impact the separation significantly. The peaks here exhibit the least amount of chromatographic "bad behavior" when compared to the other two purifications. The three labeled peaks were concentrated and evaluated by analytical HPLC, Figure 7.
The analytical only confirm what was observed during the actual purification. Interestingly, peak 1 and peak 3 contain nearly identical compositions, despite significant differences in retention. The first peak, as with the previous purifications, is attributed to the oxidized product, with some contamination by the desired peptide. Importantly though, the desired peptide is contained within peak 2 and has the highest level of purity and yield for the three samples compared herein.
I hope this data has helped to demonstrate how important the choice of solvation conditions, prior to purification can be when thinking about purification efficiency. Purification already turns into a bottleneck for most peptide teams, why do we want it to be even worse? A couple of minutes with Google, or even just thinking about the peptide in questions can help to eliminate multiple rounds of purification, or even a full re-synthesis due to poor yields.
If you would like to learn more about this and other strategies that can improve your peptide purification efficiency, click the link below.
